Amiotrofik lateral skleroz - Amyotrophic lateral sclerosis
| Amiotrofik lateral skleroz (ALS) | |
|---|---|
| Boshqa ismlar | Lou Gerig kasalligi; Charcot kasalligi; motorli neyron kasalligi (MND)[1] |
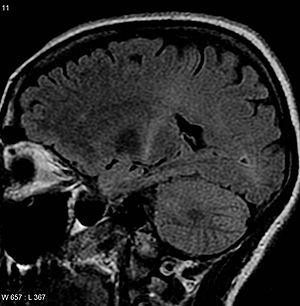 | |
| An Miyaning MRI bilan T2 signalini oshirdi orqa qismida ichki kapsula buni kuzatib borish mumkin motor korteksi, ALS tashxisiga mos keladi | |
| Mutaxassisligi | Nevrologiya |
| Alomatlar | Qattiq mushaklar, mushaklarning qisilishi, asta-sekin kuchayib borayotgan zaiflik[2] |
| Asoratlar | Qiyinchilik Gapirmoqda, yutish va nafas olish; nafas etishmovchiligi[2] |
| Odatiy boshlanish | 50-60 yillar[3] |
| Sabablari | Noma'lum (ko'p), meros qilib olingan (oz) |
| Diagnostika usuli | Alomatlar asosida gumon qilinmoqda va tomonidan qo'llab-quvvatlanmoqda MRI[2] |
| Davolash | İnvaziv bo'lmagan shamollatish[4] |
| Dori-darmon | Riluzol, edaravone[5][6] |
| Prognoz | O'rtacha umr ko'rish davomiyligi 2-4 yil[4] |
| Chastotani | Yiliga 2,6 / 100,000 (Evropa)[7] |
Amiotrofik lateral skleroz (ALS), shuningdek, nomi bilan tanilgan Lou Gerigning kasalligi Kanada va AQShda va boshqalar vosita neyron kasalligi (MND) Buyuk Britaniyada, Irlandiya, Avstraliya, Janubiy Afrika va Yangi Zelandiyada, a neyrodejenerativ asab-mushak kasalligi bu tobora yo'qotishga olib keladi vosita neyronlari bu boshqaruv ixtiyoriy mushaklar.[2][8][9] ALS - bu eng keng tarqalgan turi motorli neyron kasalligi.[10][11]ALSning dastlabki belgilariga quyidagilar kiradi qattiq mushaklar, mushaklarning qisilishi, va asta-sekin o'sib boradigan zaiflik va mushaklarning ozishi.[2] Bu ma'lum bo'lganida, qo'l yoki oyoqlarda zaiflik bilan boshlanishi mumkin oyoq-qo'llarining boshlanishiyoki qiyinchilik bilan Gapirmoqda yoki yutish sifatida tanilganida bulbar boshlanishi.[2][12] Ta'sir qilingan odamlarning taxminan yarmi kamida engil qiyinchiliklarga duch kelishadi fikrlash va xatti-harakatlar va ko'pchilik odamlar boshdan kechiradilar og'riq.[13][14] Ta'sir qilingan mushaklar ovqatni chaynash, gapirish va yurish uchun javobgardir.[2] Dvigatel neyronlarning yo'qolishi ovqatlanish, gapirish, harakatlanish va nihoyat nafas olish qobiliyati yo'qolguncha davom etadi.[2] ALS nihoyat sabab bo'ladi falaj va erta o'lim, odatda nafas etishmovchiligi.[15]
ALS kasalligining aksariyat holatlari (taxminan 90% dan 95% gacha) ma'lum bir sababga ega emas va ular sifatida ma'lum kamdan-kam uchraydigan ALS.[2][16] Ammo ikkalasi ham genetik va atrof-muhit omillari aloqador deb ishoniladi.[17] Qolgan 5% dan 10% gacha bo'lgan holatlar a bilan bog'liq bo'lgan genetik sababga ega oilada kasallik tarixi va ular quyidagicha tanilgan oilaviy ALS.[16][3] Ushbu genetik holatlarning yarmiga yaqini ikkita o'ziga xos holatga bog'liq genlar.[2] Asosiy mexanizm ikkalasiga ham zarar etkazishni o'z ichiga oladi yuqori va pastki motor neyronlari.[2] The tashxis bir kishining alomatlari va alomatlar, boshqa mumkin bo'lgan sabablarni istisno qilish uchun test sinovlari bilan.[2]
ALSni davolash mumkin emas va davolash simptomlarni yaxshilashga qaratilgan.[8] Dori chaqirildi riluzol hayotni taxminan ikki-uch oyga uzaytirishi mumkin.[5] İnvaziv bo'lmagan shamollatish hayotning yaxshilanishi va umr ko'rishiga olib kelishi mumkin.[4] Mexanik shamollatish hayotni uzaytirishi mumkin, ammo kasallikning rivojlanishini to'xtatmaydi.[18] A oziqlantirish trubkasi yordam berishi mumkin.[19] Kasallik har qanday yoshdagi odamlarga ta'sir qilishi mumkin, ammo odatda 60 yoshdan, merosxo'r hollarda esa 50 yoshdan boshlanadi.[3] Boshlanishdan o'limga qadar o'rtacha omon qolish ikki-to'rt yilni tashkil qiladi, ammo bu o'zgarishi mumkin va taxminan 10% 10 yildan ko'proq yashaydi,[4][20][2] va o'lim odatda nafas olish etishmovchiligiga bog'liq.[3] Evropada bu kasallik yiliga 100000 kishi boshiga taxminan ikki-uch kishiga ta'sir qiladi.[7] Dunyoning aksariyat qismida narxlar aniq emas.[21] Qo'shma Shtatlarda bu ko'proq tarqalgan oq tanlilar dan qora tanli odamlar.[22]
Kasallikning tavsiflari kamida 1824 yilga to'g'ri keladi Charlz Bell.[23] 1869 yilda birinchi marta simptomlar va asosiy nevrologik muammolar o'rtasidagi bog'liqlik tasvirlangan Jan-Martin Sharko, 1874 yilda bu atamani ishlatishni boshladi amiotrofik lateral skleroz.[23] Bu 20-asrda Qo'shma Shtatlarda 1939 yilda beysbol o'yinchisiga ta'sir qilganida yaxshi ma'lum bo'lgan Lou Gerig 1963 yilda tashxis qo'yilganidan keyin butun dunyo bo'ylab kosmolog Stiven Xoking.[24][25] Birinchi ALS geni 1993 yilda birinchi bo'lib topilgan hayvon modeli 1994 yilda ishlab chiqilgan.[26][27] 2014 yilda Ice Bucket Challenge Internetda virusga aylandi va bu holat haqida jamoatchilik xabardorligini oshirdi.[28]
Tasnifi
ALS a motorli neyron kasalligi, shuningdek, "motorli neyron kasalligi" deb yozilgan, bu guruh asab kasalliklari bu tanlab ta'sir qiladi vosita neyronlari, boshqaradigan hujayralar ixtiyoriy mushaklar tananing.[2] Boshqa vosita neyron kasalliklari kiradi birlamchi lateral skleroz (PLS), progressiv mushak atrofiyasi (PMA), progressiv bulbar falaj, psevdobulbar falaj va monomel amiotrofiyasi (MMA).[29]
ALS ning o'zi bir necha xil usulda tasniflanishi mumkin: kasallikning tezlashishi bilan, bu boshlanish yoshiga bog'liq; bu oilaviy yoki vaqti-vaqti bilan bo'ladimi va mintaqa birinchi bo'lib ta'sirlangan.[2] Taxminan 25% hollarda birinchi navbatda yuz, og'iz va tomoq mushaklari ta'sirlanadi, chunki qismdagi motor neyronlari miya sopi deb nomlangan medulla oblongata (ilgari "lampochka" deb nomlangan) avval pastki motorli neyronlar bilan birga o'lishni boshlaydi. Ushbu shakl "bulbar -Set ALS ". Taxminan 5% hollarda birinchi navbatda tananing magistral mushaklari ta'sir qiladi.[3] Ko'pgina hollarda kasallik tarqaladi va boshqa o'murtqa mintaqalarga ta'sir qiladi. ALS bilan kasallangan bir nechta odamda simptomlar mavjud bo'lib, ular ikkinchi mintaqaga tarqalgunga qadar kamida 12-24 oy davomida bitta o'murtqa mintaqa bilan chegaralanadi; ALSning ushbu mintaqaviy variantlari yaxshiroq prognoz bilan bog'liq.[30]
Klassik ALS, PLS va PMA

ALS ta'sirlanadigan vosita neyronlari turlari bo'yicha tasniflanishi mumkin. Odatda yoki "klassik" ALS o'z ichiga oladi yuqori motorli neyronlar miyada va pastki motor neyronlari orqa miyada.[31] Birlamchi lateral skleroz (PLS) faqat yuqori motorli neyronlarni o'z ichiga oladi va progressiv mushak atrofiyasi (PMA) faqat pastki motorli neyronlarni o'z ichiga oladi. PLS va PMA alohida kasalliklar yoki ALSning oddiy variantlari ekanligi to'g'risida munozaralar mavjud.[13]
Klassik ALS barcha ALS holatlarining taxminan 70 foizini tashkil qiladi va ularni ajratish mumkin oyoq-qo'llari bilan boshlanadigan ALS (shuningdek, orqa miya boshlanishi deb ham ataladi) va bulbar boshlangan ALS.[13] Oyoq-qo'llarida paydo bo'ladigan ALS, qo'l va oyoqlarda kuchsizlikdan boshlanadi[12] va barcha klassik ALS holatlarining uchdan ikki qismiga to'g'ri keladi.[13] Bulbar boshlangan ALS nutq, chaynash va yutish mushaklaridagi kuchsizlikdan boshlanadi[31] ishlarning qolgan uchdan bir qismini tashkil qiladi.[13] Bulbar boshlanishi, oyoq-qo'llari bilan boshlanadigan ALSga qaraganda yomon prognoz bilan bog'liq; aholiga asoslangan tadqiqot shuni ko'rsatdiki, bulbar boshlangan ALS o'rtacha omon qolish darajasi 2,0 yil va 10 yillik omon qolish darajasi 3%, oyoq-qo'llari boshlangan ALS esa o'rtacha omon qolish darajasi 2,6 yil va 10 yillik omon qolish darajasi 13 %.[32] Noyob variant - bu nafas olish yo'llari bilan boshlangan ALS, bu ALS holatlarining taxminan 3 foizini tashkil qiladi,[13] unda dastlabki alomatlar nafas olish qiyinlashadi (nafas qisilishi ) kuch bilan, dam olish paytida yoki yotgan holda (ortopnea ).[33] Umurtqa pog'onasi va bulbar simptomlari boshida yumshoq yoki yo'q bo'lib ko'rinadi. Bu erkaklarda ko'proq uchraydi.[20] Nafas olishning boshlanishi bo'lgan ALS ALS variantlarining eng yomon prognoziga ega; aholiga asoslangan tadqiqotda, nafas olish boshlanganda o'rtacha omon qolish 1,4 yil va 10 yil davomida 0% omon qolish.[32]
Birlamchi lateral skleroz (PLS) barcha ALS holatlarining taxminan 5% ni tashkil qiladi va qo'l va oyoqlarda yuqori motorli neyronlarga ta'sir qiladi.[20] Biroq, aniq PLS bilan kasallangan odamlarning 75% dan ko'prog'i simptom paydo bo'lganidan keyin to'rt yil ichida pastki motorli neyron belgilarini rivojlantiradi, ya'ni PLS aniq tashxisini shu vaqtgacha qo'yish mumkin emas.[34] PLS klassik ALSga qaraganda yaxshiroq prognozga ega, chunki u sekinroq rivojlanib, kam funktsional pasayishga olib keladi, nafas olish qobiliyatiga ta'sir qilmaydi va ozroq vazn yo'qotadi.[20]
Progressiv mushak atrofiyasi (PMA) ALSning barcha holatlarining taxminan 5% ni tashkil qiladi va qo'l va oyoqlarda pastki motorli neyronlarga ta'sir qiladi.[20] PMA klassik ALSga qaraganda o'rtacha umr ko'rish bilan bog'liq bo'lsa-da, u vaqt o'tishi bilan boshqa o'murtqa mintaqalarga o'tadi va natijada nafas olish etishmovchiligi va o'limga olib keladi.[13] Yuqori motorli neyron belgilar PMA kursining oxirida rivojlanishi mumkin, bu holda diagnostika klassik ALSga o'zgartirilishi mumkin.[34]
Mintaqaviy variantlar
ALSning mintaqaviy variantlarida kamida bir yil davomida bitta o'murtqa mintaqa bilan chegaralanadigan alomatlar mavjud; ular klassik ALSga qaraganda sekinroq rivojlanib boradi va uzoq umr ko'rish bilan bog'liq. Bunga qanot qo'llari sindromi, oyoq qanotlari sindromi va ajratilgan bulbar ALS kiradi. Flail arm sindromi va flail leg sindromi ko'pincha PMA ning mintaqaviy variantlari hisoblanadi, chunki ular faqat pastki motorli neyronlarni o'z ichiga oladi. Izolyatsiya qilingan bulbar ALS yuqori yoki pastki motor neyronlarini o'z ichiga olishi mumkin. ALSning ushbu mintaqaviy variantlarini alomatlar boshlanganda aniqlash mumkin emas; uzoq vaqt davomida (kamida 12 oy) kasallikning boshqa o'murtqa mintaqalarga tarqalishining buzilishi kuzatilishi kerak.[30]
Brakiyal amiotrofik diplegiya deb ham ataladigan qo'l suyagi sindromi,[a] faqat servikal o'murtqa miyaning pastki motorli neyron shikastlanishi bilan tavsiflanadi, bu esa proksimal qo'l mushaklaridagi zaiflikning asta-sekin paydo bo'lishiga va reflekslarning pasayishiga yoki yo'qligiga olib keladi. Oyoq amyotrofik diplegiyasi deb ham ataladigan oyoq suyagi sindromi,[b] faqat lumbosakral o'murtqa miyaning pastki motorli neyron shikastlanishi bilan tavsiflanadi, bu esa oyoqlarda zaiflikning asta-sekin paydo bo'lishiga va reflekslarning pasayishiga yoki yo'qligiga olib keladi. Izolyatsiya qilingan bulbar ALS faqat bulbar mintaqasida yuqori yoki quyi motorli neyronlarning shikastlanishi bilan tavsiflanadi, bu esa nutq bilan asta-sekin qiyinchiliklarga olib keladi (dizartriya ) va yutish (disfagiya ); nafas olish (nafas olish) odatda hech bo'lmaganda dastlab saqlanib qoladi. Ikkita kichik tadqiqotlar shuni ko'rsatdiki, izolyatsiya qilingan bulbar ALS bilan kasallanganlar, bulbar boshlangan ALS bilan kasallangan odamlarga qaraganda uzoqroq yashashi mumkin.[30]
Boshlanish yoshi
ALSni boshlanish yoshiga qarab ham tasniflash mumkin. Boshlanishning eng yuqori yoshi sporadik ALS uchun 58 dan 63 gacha, oilaviy ALS uchun 47 dan 52 gacha bo'lsa,[3] barcha ALS holatlarining taxminan 10% 45 yoshdan oldin boshlanadi ("yosh boshlangan" ALS) va barcha holatlarning taxminan 1% 25 yoshdan (voyaga etmaganlar uchun ALS) boshlanadi.[31] Yosh boshlangan ALSni rivojlantiradigan odamlarda erkaklar ko'proq bo'ladi, bulbar simptomlari kamroq uchraydi va kasallikning sekin rivojlanishi kuzatiladi.[34] Voyaga etmaganlar uchun ALS oilaviy bo'lishi ehtimoli kattalar boshlagan ALSga qaraganda ko'proq; balog'atga etmagan bolalarning ALS bilan bog'liqligi ma'lum bo'lgan genlarni o'z ichiga oladi ALS2, SETX, SPG11, FUS va SIGMAR1. Voyaga etmagan ALS bilan kasallangan odamlarning aksariyati kattalarda boshlangan ALS bilan kasallanganlarga qaraganda uzoqroq umr ko'rsalar ham, ularning ba'zilari o'ziga xos mutatsiyalarga ega FUS va SOD1 yomon prognoz bilan bog'liq.[35] Kech boshlanishi (65 yoshdan keyin) tezroq funktsional pasayish va hayotning qisqarishi bilan bog'liq.[36]
Belgilari va alomatlari
Buzilish mushaklarning zaiflashishiga olib keladi, atrofiya va mushaklarning spazmlari yuqori vosita va pastki motorli neyronlarning degeneratsiyasi tufayli butun tanada. Buzuqlikdan ta'sirlangan shaxslar oxir-oqibat barcha ixtiyoriy harakatlarni boshlash va boshqarish qobiliyatini yo'qotishi mumkin,[4] siydik pufagi va ichak faoliyati va ko'zdan tashqari mushaklar (ko'z harakati uchun mas'ul bo'lgan mushaklar) odatda tejamkor bo'ladi[37][c] kasallikning so'nggi bosqichiga qadar.[39]
Kognitiv yoki xulq-atvorining buzilishi ALS bilan og'rigan odamlarning 30-50 foizida mavjud.[40] ALS bilan kasallangan odamlarning taxminan yarmi idrok va xulq-atvorda engil o'zgarishlarni boshdan kechiradi va 10-15% da alomatlari namoyon bo'ladi frontotemporal demans.[4] Fraza yoki imo-ishoralarni takrorlash, beparvolik va inhibisyonni yo'qotish ALSning xulq-atvor xususiyatlari haqida tez-tez xabar beriladi.[41] Tilning buzilishi, ijro etuvchi funktsiya buzilishi va muammolar ijtimoiy bilish va og'zaki xotira ALSda eng ko'p uchraydigan kognitiv alomatlar; meta-tahlil disfunktsiya va kasallikning og'irligi o'rtasida hech qanday bog'liqlik topmadi.[42] Shu bilan birga, kognitiv va xulq-atvor buzilishlari ALS bilan kasallangan odamlarda omon qolish darajasining pasayishi va parvarish qiluvchi yukining ko'payishi bilan bog'liqligi aniqlandi; bunga qisman ijtimoiy bilishdagi kamchiliklar sabab bo'lishi mumkin.[42] ALS tajribasiga ega odamlarning taxminan yarmi hissiy labillik, unda ular hech qanday sababsiz yig'laydilar yoki kuladilar; bulbar boshlangan ALS bilan kasallanganlarda ko'proq uchraydi.[4]
Og'riq - bu ALS bilan kasallangan ko'pchilik odamlar boshdan kechiradigan alomatdir va bu shaklni olishi mumkin neyropatik og'riq (asab buzilishidan kelib chiqqan og'riq), spastisit, mushak kramplari va nosiseptiv og'riq harakatchanlikni pasayishi va mushaklar kuchsizligidan kelib chiqadi; ALSdagi nosiseptiv og'riq misollari kiradi kontrakturalar (mushak yoki bo'g'imning doimiy qisqarishi), bo'yin og'rig'i, bel og'rig'i, elka og'rig'i va bosim yarasi.[14]
Sensor asab va avtonom asab tizimi odatda zarar ko'rmaydi, ya'ni ALS bilan kasallangan odamlarning aksariyati eshitish, ko'rish, teginish, hid va ta'mi.[2]
Dastlabki alomatlar
ALSning boshlanishi shunchalik nozik bo'lishi mumkinki, alomatlar e'tiborga olinmaydi.[2] ALSning dastlabki belgilari mushaklarning kuchsizlanishi yoki mushak atrofiyasi. Boshqa ko'rinadigan alomatlar orasida yutish yoki nafas olish, kramp yoki ta'sirlangan mushaklarning qattiqligi; qo'l yoki oyoqqa ta'sir qiladigan mushaklar kuchsizligi; yoki noaniq va nazal nutq. ALSning dastlabki alomatlaridan ta'sirlangan tananing qismlari tanadagi qaysi vosita neyronlari birinchi navbatda zararlanishiga bog'liq.[8]
Oyoq-qo'llarida paydo bo'lgan ALSda birinchi alomatlar qo'llarda yoki oyoqlarda bo'ladi. Agar birinchi navbatda oyoqlarga ta'sir etilsa, odamlar yurish yoki yugurish paytida noqulaylik, qoqilish yoki qoqilib ketishi mumkin; bu ko'pincha "bilan yurish bilan belgilanadioyoq tashladi "Agar qo'llar birinchi navbatda ta'sirlansa, ular ko'ylakni tugmachalash, yozuv yoki qulfdagi kalitni burish kabi qo'lda epchillikni talab qiladigan ishlarda qiynalishi mumkin.[8]
Bulbar boshlangan ALSda birinchi alomatlar - gapirish yoki yutish qiyinlashadi. Nutq xiralashishi, xarakteri burun yoki tinchroq bo'lishi mumkin. Yutish va tilning harakatchanligini yo'qotish bilan bog'liq qiyinchiliklar bo'lishi mumkin. Odamlarning kichik qismi "nafas olish boshlanishi" bilan kasallanishni boshdan kechirmoqda, bu erda interkostal mushaklar birinchi navbatda nafas olishni qo'llab-quvvatlovchi ta'sir ko'rsatadi.[3]
Vaqt o'tishi bilan odamlar harakat qilishda, yutishda tobora ko'payib borayotgan qiyinchiliklarga duch kelishmoqda (disfagiya ) va so'zlash yoki so'zlarni shakllantirish (dizartriya ). Yuqori motorli neyron tutilishining alomatlari qattiq va qattiq mushaklarni o'z ichiga oladi (spastiklik ) va abartılı reflekslar (giperrefleksiya ), shu jumladan haddan tashqari faol gag refleksi. Odatda chaqiriladigan g'ayritabiiy refleks Babinskiy belgisi shuningdek, yuqori motorli neyronlarning shikastlanishini ko'rsatadi. Pastki motorli neyron degeneratsiyasining alomatlariga mushaklarning zaiflashishi va atrofiyasi, mushaklarning kramplari va mushaklarning uchib ketishi kiradi.hayratga soladigan narsalar ). Biroq, tebranish diagnostik alomatdan ko'ra ko'proq yon ta'sir qiladi; u zaiflik va atrofiyadan keyin paydo bo'ladi yoki unga hamroh bo'ladi.[2]
Taraqqiyot
Dastlabki alomatlar va rivojlanish tezligi har bir odamda turlicha bo'lishiga qaramay, kasallik oxir-oqibat ta'sirlanmagan mintaqalarga tarqaladi va ta'sirlangan hududlar ko'proq ta'sir qiladi. Aksariyat odamlar oxir-oqibat yura olmaydilar yoki qo'llari va qo'llarini ishlata olmaydilar, gapirish va ovqatni yutish qobiliyatini va o'z tupuriklarini yo'qotadilar va yo'tal va o'z-o'zidan nafas olish qobiliyatini yo'qotadilar.[4]
Progressiya tezligini. Yordamida o'lchash mumkin ALS funktsional reyting shkalasi - qayta ko'rib chiqilgan (ALSFRS-R), 12-moddadan iborat bo'lib, 48 (normal funktsiya) va 0 (og'ir nogironlik) o'rtasida ball hosil qiluvchi klinik intervyu yoki o'z-o'zidan xabar qilingan anketa sifatida o'tkaziladi;[43] bu klinik tadkikotlarda eng ko'p ishlatiladigan natija o'lchovidir va shifokorlar kasallikning rivojlanishini kuzatishda foydalanadilar.[44] O'zgaruvchanlik darajasi yuqori bo'lsa-da va odamlarning ozgina qismi ancha sekinroq tartibsizlikka ega bo'lsa-da, o'rtacha ALS bilan kasallanganlar oyiga 0,9 FRS ball yo'qotishadi. Klinisyenler o'rtasida o'tkazilgan so'rovga asoslangan tadqiqot shuni ko'rsatdiki, ular ALSFRS-R qiyaligining 20% o'zgarishini klinik jihatdan mazmunli deb baholashdi.[45]
Kasallikning kuchayishi 40 yoshdan kichik bo'lgan odamlarda sekinroq bo'ladi,[46] engil semirib ketgan,[47] birinchi navbatda bir oyoq bilan cheklangan alomatlar va asosan yuqori motorli neyron belgilari bo'lganlar.[32] Aksincha, bulbar boshlangan ALS, nafas olish yo'llari bilan boshlanadigan ALS va frontotemporal demansga chalingan odamlarda rivojlanish tezroq va prognoz yomonroq.[32]
Oxirgi bosqichlar
Chaynash va yutish bilan bog'liq qiyinchiliklar ovqatlanishni juda qiyinlashtiradi va bo'g'ilib qolish yoki ovqatni o'pkaga singdirish xavfini oshiradi. Buzilishning keyingi bosqichlarida, aspiratsion pnevmoniya rivojlanishi mumkin va sog'lom vaznni saqlash ovqatlanish trubkasini kiritishni talab qilishi mumkin bo'lgan muhim muammoga aylanishi mumkin. Diafragma sifatida va interkostal mushaklar ning ko'krak qafasi nafas olishni qo'llab-quvvatlovchi vositalar zaiflashadi o'pka funktsiyasi kabi hayotiy imkoniyatlar va ilhomlantiruvchi bosim kamayadi. Nafas olish bilan boshlangan ALSda bu oyoq-qo'llarning sezilarli darajada zaiflashishi paydo bo'lishidan oldin sodir bo'lishi mumkin. ALS bilan kasallanganlar orasida o'limning eng keng tarqalgan sababi nafas etishmovchiligi yoki zotiljam[3] va ALS bilan kasallangan odamlarning aksariyati avvalgi sababga ko'ra o'z uylarida vafot etishadi, uxlash vaqtida nafaslari to'xtaydi.[8]
Nafas olishni qo'llab-quvvatlash nafas olish bilan bog'liq muammolarni engillashtirishi va yashashni uzaytirishi mumkin bo'lsa-da, bu ALS rivojlanishiga ta'sir qilmaydi. ALS bilan kasallangan odamlarning ko'pi tashxis qo'yilganidan keyin ikki yildan to'rt yilgacha vafot etadi.[4] ALS bilan kasallangan odamlarning taxminan yarmi alomatlar boshlanganidan keyin 30 oy ichida vafot etadi va ALS bilan kasallangan odamlarning taxminan 20% alomatlar boshlangandan keyin besh yildan 10 yilgacha yashaydi.[3] Gitarachi Jeyson Beker 1989 yildan beri tartibsizlik bilan yashaydi kosmolog Stiven Xoking tashxis qo'yilganidan keyin yana 55 yil yashagan, ammo ular g'ayrioddiy holatlar hisoblanadi.[48]
Sababi
ALSning aniq sababi noma'lum bo'lsa-da, genetik va atrof-muhit omillari taxminan teng ahamiyatga ega deb o'ylashadi.[17] Genetik omillar atrof-muhit omillariga qaraganda yaxshiroq tushuniladi; hech qanday aniq ekologik omil ALSni keltirib chiqarishi aniq ko'rsatilmagan. A javobgarlik chegarasi modeli chunki ALS uyali zarar tug'ilish paytida mavjud bo'lgan genetik omillar va hayot davomida atrof-muhit xavfiga ta'sir qilish natijasida vaqt o'tishi bilan to'planib borishini taklif qiladi.[21]
Genetika
ALS kasallikning oilaviy tarixi bor yoki yo'qligiga qarab, oilaviy yoki sporadik deb tasniflanishi mumkin.[20][49] Oilaviy ALSning aniq ta'rifi bo'yicha nevrologlar o'rtasida kelishuv mavjud emas. Eng qat'iy ta'rif shuki, ALS bilan kasallangan odam ikki yoki undan ko'prog'iga ega bo'lishi kerak birinchi darajadagi qarindoshlar (bolalar, aka-ukalar yoki ota-onalar) ham ALSga ega. Kamroq aniq ta'rif shundaki, ALS bilan kasallangan odam kamida bitta birinchi darajaga ega bo'lishi kerak ikkinchi darajali nisbiy (ota-bobolari, nabiralari, xolalari, amakilari, jiyanlari, jiyanlari yoki opa-singillari) ham ALSga ega.[50] Oilaviy ALS odatda ALS kasalligining 10 foizini tashkil qiladi, deyishadi, ammo taxminlar 5 foizni tashkil qiladi[51] 20% gacha.[52] Yuqori hisob-kitoblar oilaviy ALSning kengroq ta'rifidan foydalanadi va ALS bilan kasallangan odamlarning oilaviy tarixini chuqurroq o'rganib chiqadi.[50]
Sporadik ALSda kasallikning oilaviy tarixi yo'q.[39] Sporadik ALS va oilaviy ALS klinik va patologik jihatdan bir xil ko'rinadi va genetik jihatdan o'xshashdir;[52] vaqti-vaqti bilan ALS bilan kasallangan odamlarning taxminan 10% genlarda mutatsiyaga uchraydi, ular oilaviy ALSni keltirib chiqaradi.[13] Ushbu o'xshashliklar asosida "sporadik ALS" atamasi chalg'ituvchi deb tanqid qilindi, chunki bu tasodifiy ALS holatlari faqat atrof-muhit omillaridan kelib chiqadi; aniqroq alternativa sifatida "ajratilgan ALS" atamasi taklif qilingan.[52]
20 dan ortiq genlar oilaviy ALS bilan bog'liq bo'lib, ulardan to'rttasi oilaviy holatlarning aksariyatini tashkil qiladi:[53] C9orf72 (40%), SOD1 (20%), FUS (1-5%) va TARDBP (1–5%).[13] Oilaviy ALS genetikasi sporadik ALS genetikasidan yaxshiroq tushuniladi;[13] 2016 yildan boshlab[yangilash], ma'lum ALS genlari oilaviy ALSning taxminan 70 foizini va sporadik ALSning taxminan 15 foizini tushuntirdi.[54][55] Umuman olganda, ALS bilan kasallangan shaxsning birinchi darajali qarindoshlarida ALS rivojlanish xavfi 1% ni tashkil qiladi.[17][56] ALS an merosning oligogenik usuli, ya'ni kasallik keltirib chiqarishi uchun ikki yoki undan ortiq gendagi mutatsiyalar talab qilinadi.[26]
ALS va frontotemporal demans (FTD) hozirgi kunda genetik, klinik va patologik o'xshashlik tufayli keng tarqalgan kasallik spektrining (FTD-ALS) bir qismi hisoblanadi.[57] Genetik jihatdan, C9orf72 takroriy kengayishlar oilaviy ALSning taxminan 40% va oilaviy FTDning 25% ni tashkil qiladi.[26] Klinik jihatdan, ALS bilan kasallangan odamlarning 50% ba'zi bir bilim yoki xulq-atvor buzilishlariga ega va 5-15% FTDga ega, FTD bilan kasallanganlarning 40% ba'zi motorli neyron belgilariga ega va 12,5% ALSga ega.[13] Patologik jihatdan TDP-43 oqsilining g'ayritabiiy agregatlari ALS bemorlarining 97 foizigacha va FTD bemorlarining 50 foizigacha kuzatiladi.[58] FTD-ALS ni keltirib chiqarishi ma'lum bo'lgan boshqa genlarga kiradi CHCHD10, SQSTM1 va TBK1.[53]
Atrof-muhit omillari
Kasallikning oilaviy tarixi bo'lmagan joylarda - taxminan 90% holatlar - sabablari ma'lum emas. Dalillar aniq bo'lmagan mumkin bo'lgan uyushmalarga harbiy xizmat va chekish kiradi.[40] Harbiy tarix va ALS chastotasi bo'yicha tadqiqotlar bir-biriga zid bo'lsa-da, a uchun zaif dalillar mavjud ijobiy korrelyatsiya.[59] Turli xil taklif etilayotgan omillarga ta'sir qilish kiradi atrof-muhit toksinlari (geografik joylashishni o'rganish bo'yicha xulosalar), shuningdek, harbiy xizmat paytida alkogol va tamaki iste'mol qilish.[59]
2016 yilda o'tkazilgan 16 ta meta-tahlilni o'rganish natijasida kasbiy surunkali ta'sir qilish bilan bog'liq bo'lgan ishonchli dalillar mavjud degan xulosaga kelishdi qo'rg'oshin; dehqonchilik, qo'rg'oshin, beta-karotin iste'mol qilish va bosh jarohati tashqari og'ir metallarga ta'sir qilish uchun dalillar; va omega-uch yog 'kislotasini iste'mol qilish, o'ta past chastotali elektromagnit maydonlar, zararkunandalarga qarshi vositalar va siydik kislotasi ta'siriga oid zaif dalillar.[60]
Qo'shma Shtatlar tomonidan 2017 yilda o'tkazilgan tadqiqotda Kasalliklarni nazorat qilish va oldini olish markazlari 1985 yildan 2011 yilgacha bo'lgan AQSh o'limini tahlil qilib, ALS o'limi bilan bog'liq kasblar oq yoqalilar masalan, menejment, moliyaviy, me'moriy, hisoblash, yuridik va ta'lim ishlarida.[61] Boshqa potentsial xavf omillari tasdiqlanmagan bo'lib qolmoqda, jumladan kimyoviy ta'sir, elektromagnit maydonga ta'sir qilish, ishg'ol qilish, jismoniy shikastlanish va elektr toki urishi.[62][63] Turli xil ta'sir qilish bilan taxminiy bog'liqlik mavjud pestitsidlar shu jumladan xlor organik hasharotlar aldrin, dieldrin, DDT va toksafen.[64][65][66]
Bosh jarohati
2015 yilgi tekshiruv o'rtacha va og'ir ekanligini aniqladi shikast miya shikastlanishi ALS uchun xavf omilidir, ammo miyaning engil shikastlanish darajasi oshishi aniq emas edi.[67] 2017 yilgi meta-tahlil natijasida bosh jarohati va ALS o'rtasidagi bog'liqlik aniqlandi; ammo, mualliflar teskari sabablarni keltirib chiqarish imkoniyatini ko'rib chiqqanlarida, bu assotsiatsiya g'oyib bo'ldi, ya'ni bu bosh jarohatlari ALSning sababi emas, balki aniqlanmagan ALSning dastlabki alomati.[68]
Jismoniy faoliyat
Bir qator sharhlar jismoniy faollik miqdori va ALS rivojlanish xavfi o'rtasida hech qanday bog'liqlik topmadi.[69][70][71] 2009 yilgi tadqiqotlar shuni ko'rsatdiki, jismoniy faoliyatning ALS uchun xavf omili sifatida cheklangan, ziddiyatli va qat'iy xulosaga kelish uchun sifat etarli emas.[72] 2014 yilgi xulosada jismoniy faoliyat umuman ALS uchun xavf tug'dirmaydi, futbol va Amerika futboli ehtimol ALS bilan bog'liqligi va jismoniy talabchan kasblarning ALS bilan bog'liqligini aytish uchun etarli dalillar yo'q degan xulosaga kelishdi.[73] 2016 yilgi tekshiruv dalillarni xulosasiz deb topdi va tadqiqotlarning dizaynidagi farqlar tadqiqotlarni taqqoslashni qiyinlashtirayotganini ta'kidladi, chunki ular bir xil jismoniy faollik yoki ALS uchun bir xil diagnostik mezonlardan foydalanmaydi.[74]
Sport
Ham futbol, ham Amerika futboli bir nechta tadqiqotlarda ALS uchun xavf omillari sifatida aniqlangan, ammo bu assotsiatsiya kam sonli ALS holatlariga asoslangan.[75] 2012 yilgi 3439 ta retrospektiv kohort tadqiqotlari NFL futbolchilar neyrodejenerativ sabablarga ko'ra o'lish xavfi AQShning umumiy aholisidan uch baravar yuqori va ALS yoki Altsgeymer kasalligidan o'lish xavfi to'rt baravar yuqori ekanligini aniqladilar.[76] Shu bilan birga, ushbu xavf Altsgeymer kasalligidan ikki o'lim va ushbu kohortdagi jami 334 o'limdan oltita o'lim asosida hisoblab chiqilgan, ya'ni ushbu tadqiqot Amerika futbolini o'ynash ALS uchun xavfli omil ekanligini aniq isbotlamaydi.[77] ALSdan vafot etgan deb o'ylagan ba'zi NFL o'yinchilari aslida bo'lishi mumkin edi surunkali shikastli ensefalopatiya (CTE), ALSga juda o'xshash alomatlar ko'rsatishi mumkin bo'lgan ko'plab bosh jarohatlari bilan bog'liq bo'lgan neyrodejenerativ kasallik.[67][d]
1960 yildan 1996 yilgacha o'ynagan 24000 italiyalik futbolchini retrospektiv ravishda olib borgan kohort tadqiqotida futbol ALS uchun xavfli omil sifatida aniqlandi. Ushbu guruhda 375 kishi o'lgan, shu jumladan sakkiz nafari ALSdan. Ushbu ma'lumotlarga va ALS bilan kasallanish holatlariga asoslanib, futbolchilar umumiy Italiya aholisiga qaraganda ALSdan o'lish ehtimoli 11 baravar ko'p ekanligi aniqlandi.[21] Biroq, ushbu hisob-kitob kogortada ALSning kutilgan kam sonli holatlariga tayanishi uchun tanqid qilindi.[72] Kutilgan holatlar sonini taxmin qilish uchun ALS rivojlanishining umr bo'yi xavfi ishlatilganda, futbolchilar umumiy populyatsiyadan ko'ra ALSdan o'lish ehtimoli yo'q edi.[21]
Chekish
Chekish, ehtimol ALS bilan bog'liq. 2009 yilgi tekshiruv natijalariga ko'ra, chekish ALS uchun xavf omilidir.[80] 2010 yilgi muntazam tekshiruv va meta-tahlil natijalariga ko'ra, chekish va ALS o'rtasida kuchli bog'liqlik yo'q, ammo chekish ayollarda ALS xavfi yuqori bo'lishi mumkin.[81] 2011 yilgi meta-tahlil natijalariga ko'ra, chekish ALS xavfini oshiradi va hech qachon chekmaydi. Chekuvchilar orasida yoshroq chekishni boshlaganlarida, ular ALS bilan kasallanish ehtimoli ko'proq bo'lgan; ammo, chekilgan yillar soni ham, kuniga chekilgan sigaretalar soni ham ularning ALS rivojlanish xavfiga ta'sir qilmadi.[82]
Patofiziologiya
Neyropatologiya
ALSni belgilovchi xususiyati ikkala yuqori motorli neyronlarning o'limidir motor korteksi miya) va pastki motorli neyronlar (miya sopi va orqa miyada joylashgan).[83] Frontotemporal demans bilan og'rigan ALSda miyaning frontal va temporal loblari bo'ylab neyronlar ham nobud bo'ladi.[39] ALSning patologik alomati - bu mavjudligi inklyuziya organlari (oqsilning g'ayritabiiy agregatlari) sifatida tanilgan Bunina tanalari motorli neyronlarning sitoplazmasida. ALS bilan kasallangan odamlarning taxminan 97 foizida inklyuziya organlarining asosiy tarkibiy qismi hisoblanadi TDP-43 oqsil;[12] ammo, ularda SOD1 yoki FUS mutatsiyalar, inklyuziya organlarining asosiy tarkibiy qismi[84][85] navbati bilan SOD1 oqsili yoki FUS oqsilidir.[31] The yalpi patologiya Yalang'och ko'z bilan ko'rish mumkin bo'lgan kasallikning o'ziga xos xususiyati bo'lgan ALSga skelet mushaklari atrofiyasi, motor korteks atrofiyasi, skleroz kiradi. kortikospinal va kortikobulbar yo'llari, ning ingichkalashi gipoglossal nervlar (tilni boshqaradigan) va orqa miyaning oldingi ildizlarini yupqalash.[12] Dvigatel neyronlarning o'limidan tashqari, ALSning ko'pgina variantlariga xos bo'lgan yana ikkita xususiyat - bu fokusli boshlang'ich patologiya, ya'ni simptomlar bitta o'murtqa mintaqada boshlanadi va doimiy ravishda doimiy tarqalib boradi, ya'ni vaqt o'tishi bilan simptomlar qo'shimcha hududlarga tarqaladi. Prion - noto'g'ri katlanmış oqsillarni hujayradan hujayraga tarqalishi kabi, ALS nima uchun bir sohada boshlanib, boshqalarga tarqalishini tushuntirishi mumkin.[31] The glimfatik tizim da ishtirok etishi mumkin patogenez ALS.[86]
Biokimyo

ALSda nega neyronlarning o'lishi hali ham to'liq tushunilmagan, ammo bu neyrodejeneratsiya turli xil uyali va molekulyar jarayonlarni o'z ichiga oladi deb o'ylashadi.[13] ALSga aloqadorligi ma'lum bo'lgan genlarni normal ishlashiga qarab uchta umumiy toifaga ajratish mumkin: oqsillarning parchalanishi, sitoskelet va RNKni qayta ishlash. Mutant SOD1 oqsillari hujayra ichidagi agregatlarni hosil qiladi, ular oqsilning parchalanishini inhibe qiladi. Ning sitoplazmatik agregatlari yovvoyi tip (normal) SOD1 oqsili sporadik ALSda keng tarqalgan.[39] Noto'g'ri katlanmış mutant SOD1 prionga o'xshash tarzda qo'shni neyronlarda yovvoyi SOD1 tipidagi noto'g'ri birikma va agregatsiyaga olib kelishi mumkin deb o'ylashadi.[12] Mutatsiyaga uchraganda ALSga olib kelishi mumkin bo'lgan boshqa protein degradatsiyasi genlari kiradi VCP, OPTN, TBK1va SQSTM1. Sitoskeletonni saqlash uchun muhim bo'lgan ALSga tegishli uchta gen[39] va aksonal transport uchun[12] o'z ichiga oladi DCTN1, PFN1 va TUBA4A.[39]
RNK bilan bog'langan oqsillarni kodlaydigan bir qator ALS genlari mavjud. Birinchisi, TDP-43 oqsili,[39] ALSning deyarli barcha holatlarida motorli neyronlarning sitoplazmasida to'planadigan yadro oqsili; ammo, mutatsiyalar TARDBP, TDP-43 uchun kod beradigan gen, ALSning kam uchraydigan sababidir.[12] FUS mutatsiyaga uchraganda ALS ni keltirib chiqarishi mumkin bo'lgan TDP-43 ga o'xshash funktsiyaga ega bo'lgan boshqa RNK-bog'lovchi oqsil FUS uchun kodlar.[26] Mutatsiyalar TARDBP va FUS murakkabligi past bo'lgan domenning bog'lanish yaqinligini oshirib, ularning tegishli oqsillarini sitoplazmada to'planishiga olib keladi. Ushbu mutant RNK-biriktiruvchi oqsillar noto'g'ri biriktirilib, to'plangandan so'ng, ular hujayralar ichida ham, hujayralar orasida ham normal oqsilni prionga o'xshash tarzda noto'g'ri qo'shib olishlari mumkin.[39] Bu shuningdek, yadroda RNK bilan bog'langan oqsil darajasining pasayishiga olib keladi, bu ularning maqsadli RNK transkriptlari normal ishlovdan o'tmasligini anglatishi mumkin. ALS bilan bog'liq boshqa RNK metabolizm genlari kiradi ANG, SETX va MATR3.[12]
C9orf72 ALSdagi eng ko'p mutatsiyaga uchragan gen bo'lib, bir qator mexanizmlar orqali motorli neyronlarning o'limiga sabab bo'ladi.[39] Patogen mutatsiya geksanukleotidning takroriy kengayishidir (oltita nukleotidlar ketma-ket takrorlanadi);[58] 30 takroriy odam normal holat, yuzlab yoki minglab takroriy odamlarda oilaviy ALS, frontotemporal demans yoki ba'zan sporadik ALS bo'lishi mumkin. Ular bilan bog'liq kasallikning uchta mexanizmi C9orf72 takroriy takrorlash - bu RNK transkriptlarining yadroda yotishi, RNKning sitoplazmadagi toksik dipeptidli takroriy oqsillarga aylanishi va normal C9orf72 oqsilining pasayishi.[39]
Eksitotoksiklik, yoki qo'zg'atuvchi nörotransmitter tomonidan haddan tashqari stimulyatsiya tufayli hujayralardagi kaltsiyning yuqori darajasidan kelib chiqqan asab hujayralarining o'limi glutamat, ALSning barcha shakllari uchun umumiy bo'lgan mexanizmdir. Dvigatel neyronlari boshqa neyronlarga qaraganda eksitotoksikaga sezgir, chunki ular kaltsiy tamponlash qobiliyatiga ega va glutamat retseptorlari turiga ega ( AMPA retseptorlari ) bu kaltsiy uchun ko'proq o'tkazuvchan. ALSda qo'zg'atuvchi aminokislota tashuvchisi 2 (EAAT2 ), bu glutamatni sinapsdan olib tashlaydigan asosiy transport vositasi; bu sinaptik glutamat darajasining oshishiga va eksitotoksiklikka olib keladi. ALSda yashashni o'rtacha darajada uzaytiradigan Riluzol preparati sinaptikgacha bo'lgan neyronlardan glutamat ajralishini inhibe qiladi; ammo, ushbu mexanizm uning terapevtik ta'siri uchun javobgar ekanligi aniq emas.[12]
Tashxis

Hech qanday test ALSning aniq tashxisini taqdim eta olmaydi, garchi bitta a'zoda yuqori va quyi motorli neyron belgilarining mavjudligi qat'iy dalolat beradi.[2] Buning o'rniga, ALS diagnostikasi birinchi navbatda alomatlarga va belgilarga asoslangan shifokor odamda va boshqa kasalliklarni istisno qilish uchun bir qator testlarni kuzatadi.[2] Shifokorlar odamni to'liq olishadi kasallik tarixi va odatda muntazam ravishda nevrologik tekshiruv o'tkazib, mushaklarning kuchsizligi, mushaklarning atrofiyasi, giperrefleksiya va spastiklik yomonlashmoqda.[2] Bir qator biomarkerlar ushbu holat bo'yicha o'rganilmoqda, ammo hozircha umumiy tibbiy foydalanishda emas.[88][89]
Diagnostika mezonlari
ALS diagnostikasi El Escorial Revised mezonlari va Avaji mezonlariga asoslanadi.[12] Dastlabki El Escorial mezonlari to'rtta o'murtqa mintaqaning: bulbar, bachadon bo'yni, torakal va bel qismlarining qancha qismini qamrab olganiga qarab diagnostik aniqlik darajasiga ega edi. Belgilangan ALS uchta o'murtqa mintaqada yuqori motorli neyron (UMN) va pastki motorli neyron (LMN) belgilari, ikkita mintaqada UMN va LMN belgilari bo'lishi mumkin bo'lgan ALS, faqat bitta mintaqada UMN va LMN belgilari bo'lishi mumkin bo'lgan shubha bilan aniqlandi ALS faqat LMN belgisi sifatida. "El Escorial Revised" mezonlari, shuningdek, "Airlie House" mezonlari deb nomlanib, "shubhali ALS" toifasini tashladi va "laboratoriya tomonidan qo'llab-quvvatlanadigan ehtimoliy ALS" toifasini qo'shdi. Awaji mezonlari g'ayritabiiy EMG testlarini ALS diagnostikasini o'tkazishda LMN disfunktsiyasining klinik belgilari bilan bir xil og'irlikni beradi,[34] shuning uchun "laboratoriya tomonidan qo'llab-quvvatlanadigan ehtimoliy ALS" toifasini keraksiz holga keltirish. Avaji mezonidagi uchta toifalar aniq ALS, ehtimoliy ALS va mumkin bo'lgan ALS.[90]
El Escorial Qayta ko'rib chiqilgan mezonlari ALS uchun xosdir, ya'ni mezonlarga javob beradigan odamda ALS bo'lishi ehtimoli yuqori; ammo, ular ALS uchun ayniqsa sezgir emaslar, ya'ni mezonlarga javob bermaydigan odam hali ham ALS kasalligiga chalingan bo'lishi mumkin. ALSning dastlabki bosqichlarida ularning sezgirligi ayniqsa yomon. Awaji mezonlari El Escorial Revised mezonlariga qaraganda, ayniqsa, bulbar boshlangan ALS uchun yaxshiroq sezgirlikka ega.[34] 2012 yilgi meta-tahlil natijalariga ko'ra El Escorial Revised mezonlari sezgirligi 62,2% ni, Awaji mezonlari esa sezgirligi 81,1% ni tashkil etgan; ikkala mezon to'plamining o'ziga xos xususiyati taxminan 98% ni tashkil etdi.[91] El Escorial mezonlari klinik sinovlar uchun bemor guruhlarini standartlashtirish uchun ishlab chiqilgan[92] ammo klinik amaliyotda unchalik foydali emas; El Escorial mezonlari bilan tavsiflangan mumkin bo'lgan ALS deyarli har doim klinik jihatdan ALS hisoblanadi.[12]
Differentsial diagnostika
ALS alomatlari turli xil, ko'proq davolanadigan kasalliklar yoki kasalliklarga o'xshash bo'lishi mumkinligi sababli, boshqa holatlar mavjudligini istisno qilish uchun tegishli testlarni o'tkazish kerak. Ushbu testlardan biri elektromiyografiya (EMG), mushaklarda elektr faolligini aniqlaydigan maxsus ro'yxatga olish texnikasi. Ba'zi EMG topilmalari ALS diagnostikasini qo'llab-quvvatlashi mumkin. Boshqa keng tarqalgan sinov choralari asabni o'tkazish tezligi NCV natijalaridagi o'ziga xos anormalliklar, masalan, odamning bir shaklga ega ekanligini ko'rsatishi mumkin. periferik neyropatiya (periferik nervlarning shikastlanishi) yoki miyopatiya ALSdan ko'ra (mushak kasalligi). A magnit-rezonans tomografiya ALSning dastlabki bosqichida bo'lgan odamlarda (MRI) odatda normal holat bo'lib, u simptomlarni keltirib chiqarishi mumkin bo'lgan boshqa muammolarning dalillarini, masalan, orqa miya shishi, skleroz, a churrasi bo'lgan disk bo'ynida, siringomiyeliya yoki bachadon bo'yni spondiloz.[2]
Shaxsning alomatlari va tekshiruvdan va ushbu testlardan olingan xulosalarga asoslanib, shifokor qon va siydik samples to eliminate the possibility of other diseases, as well as routine laboratory tests. In some cases, for example, if a physician suspects the person may have a myopathy rather than ALS, a muscle biopsy may be performed.[2]
A number of infectious diseases can sometimes cause ALS-like symptoms,[2] including human immunodeficiency virus (OIV ), inson T-limfotrop virusi (HTLV), Lyme kasalligi va sifiliz.[13] Neurological disorders such as multiple sclerosis, poliomiyelitdan keyingi sindrom, multifocal motor neuropathy, CIDP, o'murtqa mushak atrofiyasi va orqa miya va bulbar mushak atrofiyasi can also mimic certain aspects of the disease and should be considered.[2]
ALS must be differentiated from the "ALS mimic syndromes", which are unrelated disorders that may have a similar presentation and clinical features to ALS or its variants.[93] Because of the prognosis carried by this diagnosis and the variety of diseases or disorders that can resemble ALS in the early stages of the disease, people with ALS symptoms should always obtain a specialist neurological opinion in order to rule out alternative diagnoses. Myasthenic syndrome, also known as Lambert–Eaton syndrome, can mimic ALS, and its initial presentation can be similar to that of myasteniya gravis (MG), a treatable autoimmune disease sometimes mistaken for ALS.[94][95] Benign fasciculation syndrome is another condition that mimics some of the early symptoms of ALS, but is accompanied by normal EMG readings and no major disablement.[96]
Most cases of ALS, however, are correctly diagnosed, with the error rate of diagnosis in large ALS clinics being less than 10%.[97][98] One study examined 190 people who met the MND/ALS diagnostic criteria, complemented with laboratory research in compliance with both research protocols and regular monitoring. Thirty of these people (16%) had their diagnosis completely changed during the clinical observation development period.[99] In the same study, three people had a false negative diagnosis of MG, which can mimic ALS and other neurological disorders, leading to a delay in diagnosis and treatment. MG is eminently treatable; ALS is not.[100]
Menejment
There is no cure for ALS. Management focuses on treating symptoms and providing supportive care, with the goal of improving quality of life and prolonging survival.[13] This care is best provided by multidisciplinary teams of healthcare professionals; attending a multidisciplinary ALS clinic is associated with longer survival, fewer hospitalizations, and improved quality of life.[4] Riluzol prolongs survival by about 2–3 months.[5] Edaravone slows functional decline slightly in a small number of people with ALS;[101] it is expensive and must be administered by daily IV infusions that may decrease quality of life.[102] Other medications may be used to manage other symptoms.[103]
İnvaziv bo'lmagan shamollatish (NIV) is the main treatment for respiratory failure in ALS.[12] In people with normal bulbar function, it prolongs survival by about seven months and improves quality of life. One study found that NIV is ineffective for people with poor bulbar function[104] while another suggested that it may provide a modest survival benefit.[13] Many people with ALS have difficulty tolerating NIV.[105] Invasive ventilation is an option for people with advanced ALS when NIV is not enough to manage their symptoms.[4] While invasive ventilation prolongs survival, disease progression and functional decline continue.[18] It may decrease the quality of life of people with ALS or their caregivers.[19][18] Invasive ventilation is more commonly used in Japan than North America or Europe.[106]
Physical therapy can promote functional independence[107][108] through aerobic, range of motion, and stretching exercises.[103] Occupational therapy can assist with activities of daily living through adaptive equipment.[109] Speech therapy can assist people with ALS who have difficulty speaking.[108] Preventing weight loss and malnutrition in people with ALS improves both survival and quality of life.[13] Initially, difficulty swallowing (dysphagia) can be managed by dietary changes and swallowing techniques. A oziqlantirish trubkasi should be considered if someone with ALS loses 5% or more of their body weight or if they cannot safely swallow food and water.[12] The feeding tube is usually inserted by percutaneous endoscopic gastrostomy (PEG). There is weak evidence that PEG tubes improve survival.[110] PEG insertion is usually performed with the intent of improving quality of life.[19]
Palliative care should begin shortly after someone is diagnosed with ALS.[111] Discussion of end-of-life issues gives people with ALS time to reflect on their preferences for end-of-life care and can help avoid unwanted interventions or procedures. Hospice care can improve symptom management at the end of life and increases the likelihood of a peaceful death.[19] In the final days of life, opioids can be used to treat pain and dyspnea, while benzodiazepines can be used to treat anxiety.[18]
Dori vositalari

Riluzol has been found to modestly prolong survival by about 2–3 months.[112][5] It may have a greater survival benefit for those with bulbar-onset ALS.[5] It may work by decreasing release of the excitatory neurotransmitter glutamat from pre-synaptic neurons.[12] The most common side effects are nausea and a lack of energy (asteniya ).[5] People with ALS should begin treatment with riluzole as soon as possible following their diagnosis.[111]
Edaravone has been shown to modestly slow the decline in function in a small group of people with early-stage ALS.[e][f][101][114] It may work by protecting motor neurons from oksidlovchi stress.[115] The most common side effects are bruising and gait disturbance.[114] Treatment with edaravone is expensive and requires daily hour-long IV infusions for 10 days in a two-week period.[102]
Other medications may be used to help reduce fatigue, ease muscle cramps, control spasticity, and reduce excess saliva and balg'am.[103] Gabapentin, pregabalin va trisiklik antidepressantlar (masalan, amitriptilin ) can be used for neuropathic pain, while nonsteroidal anti-inflammatory drugs (NSAID ), asetaminofen va opioidlar can be used for nociceptive pain.[14]
Depression can be treated with serotoninni qaytarib olishning selektiv inhibitörleri (SSRIs) or tricyclic antidepressants,[12] esa benzodiazepinlar can be used for anxiety.[4] There are no medications to treat cognitive impairment/frontotemporal dementia (FTD); however, SSRIs and antipsychotics can help treat some of the symptoms of FTD.[12] Baklofen va tizanidine are the most commonly used oral drugs for treating spasticity; an intratekal baclofen pump can be used for severe spasticity.[12] Atropin, skopolamin, amitriptyline or glycopyrrolate may be prescribed when people with ALS begin having trouble swallowing their saliva (sialorrhea ).[12]
A 2017 review concluded that mexiletin was safe and effective for treating cramps in ALS based on a randomized controlled trial from 2016.[114] In a study from 2020, AMX0035, a combination of sodium phenylbutyrate va taurursodiol, was shown to prolong the survival of patients by several months.[116][117]
Breathing support
İnvaziv bo'lmagan shamollatish

İnvaziv bo'lmagan shamollatish (NIV) is the primary treatment for respiratory failure in ALS[12] and was the first treatment shown to improve both survival and quality of life.[4] NIV uses a face or nasal mask connected to a ventilator that provides intermittent positive pressure to support breathing. Continuous positive pressure is not recommended for people with ALS because it makes breathing more difficult.[18] Initially, NIV is used only at night[4] because the first sign of respiratory failure is decreased gas exchange (gipoventiliya ) during sleep; symptoms associated with this nocturnal hypoventilation include interrupted sleep, anxiety, morning headaches, and daytime fatigue. As the disease progresses, people with ALS develop shortness of breath when lying down, during physical activity or talking, and eventually at rest.[118] Other symptoms include poor concentration, poor memory, confusion, respiratory tract infections, and a weak cough. Respiratory failure is the most common cause of death in ALS.[4]
It is important to monitor the respiratory function of people with ALS every three months, because beginning NIV soon after the start of respiratory symptoms is associated with increased survival. This involves asking the person with ALS if they have any respiratory symptoms and measuring their respiratory function.[4] The most commonly used measurement is upright majburiy hayotiy imkoniyatlar (FVC), but it is a poor detector of early respiratory failure and is not a good choice for those with bulbar symptoms, as they have difficulty maintaining a tight seal around the mouthpiece. Measuring FVC while the person is lying on their back (supine FVC) is a more accurate measure of diaphragm weakness than upright FVC.[105] Sniff nasal inspiratory pressure (SNIP) is a rapid, convenient test of diaphragm strength that is not affected by bulbar muscle weakness.[18] If someone with ALS has signs and symptoms of respiratory failure, they should undergo daytime blood gas analysis[4] to look for gipoksemiya (low oxygen in the blood) and giperkapniya (too much carbon dioxide in the blood).[18] If their daytime blood gas analysis is normal, they should then have nocturnal impuls oksimetriyasi to look for hypoxemia during sleep.[4]
Non-invasive ventilation prolongs survival longer than riluzole. A 2006 randomized controlled trial found that NIV prolongs survival by about 48 days and improves quality of life; however, it also found that some people with ALS benefit more from this intervention than others. For those with normal or only moderately impaired bulbar function, NIV prolongs survival by about seven months and significantly improves quality of life. For those with poor bulbar function, NIV neither prolongs survival nor improves quality of life, though it does improve some sleep-related symptoms.[104] Despite the clear benefits of NIV, about 25–30% of all people with ALS are unable to tolerate it, especially those with cognitive impairment or bulbar dysfunction.[105] Results from a large 2015 cohort study suggest that NIV may prolong survival in those with bulbar weakness, and so NIV should be offered to all people with ALS, even if it is likely that they will have difficulty tolerating it.[13]
Invasive ventilation
Invasive ventilation bypasses the nose and mouth (the upper airways) by making a cut in the trachea (traxeostomiya ) and inserting a naycha connected to a ventilator.[18] It is an option for people with advanced ALS whose respiratory symptoms are poorly managed despite continuous NIV use.[4] While invasive ventilation prolongs survival, especially for those younger than 60, it does not treat the underlying neurodegenerative process. The person with ALS will continue to lose motor function, making communication increasingly difficult and sometimes leading to qulflangan sindrom, in which they are completely paralyzed except for their eye muscles.[18] About half of the people with ALS who choose to undergo invasive ventilation report a decrease in their quality of life[19] but most still consider it to be satisfactory. However, invasive ventilation imposes a heavy burden on caregivers and may decrease their quality of life.[18] Attitudes toward invasive ventilation vary from country to country; about 30% of people with ALS in Japan choose invasive ventilation, versus less than 5% in North America and Europe.[106]
Terapiya

Jismoniy davolash plays a large role in rehabilitation for individuals with ALS. Specifically, physical, occupational, and speech therapists can set goals and promote benefits for individuals with ALS by delaying loss of strength, maintaining endurance, limiting pain, improving speech and swallowing, preventing complications, and promoting functional independence.[107][108]
Occupational therapy and special equipment such as yordamchi texnologiya can also enhance people's independence and safety throughout the course of ALS.[109] Gentle, low-impact aerob mashqlari such as performing activities of daily living, walking, swimming, and stationary bicycling can strengthen unaffected muscles, improve cardiovascular health, and help people fight fatigue and depression. Range of motion and stretching exercises can help prevent painful spastiklik and shortening (contracture) of muscles. Physical and occupational therapists can recommend exercises that provide these benefits without overworking muscles, because muscle exhaustion can lead to worsening of symptoms associated with ALS, rather than providing help to people with ALS.[103] They can suggest devices such as ramps, braces, walkers, bathroom equipment (shower chairs, toilet risers, etc.), and wheelchairs that help people remain mobile. Occupational therapists can provide or recommend equipment and adaptations to enable ALS people to retain as much safety and independence in activities of daily living as possible.[109]
People with ALS who have difficulty speaking or swallowing may benefit from working with a defektolog.[108] These health professionals can teach people adaptive strategies such as techniques to help them speak louder and more clearly. As ALS progresses, speech-language pathologists can recommend the use of kuchaytiruvchi va muqobil aloqa such as voice amplifiers, speech-generating devices (or voice output communication devices) or low-tech communication techniques such as head mounted laser pointers, alphabet boards or yes/no signals.[108] Speech-language pathologists may also help people diagnosed with ALS with their swallowing impairment (dysphagia) which may include modified diet, swallowing exercises, compensatory strategies. People with ALS might require tracheostomy placement, which SLPs will help to manage.[iqtibos kerak ]
Oziqlanish

Preventing weight loss and malnutrition in people with ALS improves both survival and quality of life.[13] Weight loss in ALS is caused by muscle wasting due to motor neuron death, increased resting energy expenditure, and decreased food intake. Yutish qiyin (disfagiya ) develops in about 85% of people with ALS at some point over the course of their disease and is a major cause of decreased food intake, leading to malnutrition and weight loss.[18] It is important to regularly assess the weight and swallowing ability of people with ALS.[4] Initially, dysphagia may be managed by dietary changes and modified swallowing techniques.[12] Difficulty swallowing liquids usually develops first and can be managed by switching to thicker liquids like fruit nectar or smoothies, or by adding fluid thickeners to thin fluids like water and coffee. People with ALS should eat soft, moist foods, which tend to be easier to swallow than dry, crumbly, or chewy foods.[118] They should also be instructed on proper head posture during swallowing, which can make swallowing easier.[12] There is tentative evidence that high-calorie diets may prevent further weight loss and improve survival.[114] Patients will receive speech therapy to address their dysphagia and to continuously assess for the most least restrictive, and safe diet consistency.
A oziqlantirish trubkasi should be considered if someone with ALS loses 5% or more of their body weight or if they cannot safely swallow food and water.[12] This can take the form of a gastrostomy tube, in which a tube is placed through the wall of the abdomen into the stomach, or a nazogastrik naycha, in which a tube is placed through the nose and down the esophagus into the stomach.[18] A gastrostomy tube is more appropriate for long-term use[4] than a nasogastric tube, which is uncomfortable and can cause esophageal ulcers.[18] The feeding tube is usually inserted by percutaneous endoscopic gastrostomy (PEG). There is some evidence that a PEG tube should be inserted before vital capacity drops below 50% of expected, as a low vital capacity may be associated with a higher risk of complications. However, a large 2015 study showed that PEG insertion is safe in people with advanced ALS and low vital capacities, as long as they are on NIV during the procedure.[114]
There is weak evidence that PEG tubes improve survival.[110] PEG insertion is usually performed with the intent of improving quality of life[19] by sustaining nutrition and medication intake.[4] This reduces the risk of weight loss and dehydration, and can decrease anxiety from extended mealtimes[19] and decreased oral food intake.[4]
Hayot tugashi bilan parvarish qilish
Palyativ yordam, which relieves symptoms and improves quality of life without treating the underlying disease, should begin shortly after someone is diagnosed with ALS.[111] Early discussion of end-of-life issues gives people with ALS time to reflect on their preferences for end-of-life care and can help avoid unwanted interventions or procedures.[19] Once they have been fully informed about all aspects of various life-prolonging measures, they can fill out advanced directives indicating their attitude toward noninvasive ventilation, invasive ventilation, and feeding tubes.[114] Late in the disease course, difficulty speaking due to muscle weakness (dizartriya ) and cognitive dysfunction may impair their ability to communicate their wishes regarding care.[12] Continued failure to solicit the preferences of the person with ALS may lead to unplanned and potentially unwanted emergency interventions, such as invasive ventilation. If people with ALS or their family members are reluctant to discuss end-of-life issues, it may be useful to use the introduction of gastrostomy or noninvasive ventilation as an opportunity to bring up the subject.[19]
Xospisga g'amxo'rlik, or palliative care at the end of life, is especially important in ALS because it helps to optimize the management of symptoms and increases the likelihood of a peaceful death.[19] It is unclear exactly when the end-of-life phase begins in ALS, but it is associated with significant difficulty moving, communicating, and, in some cases, thinking.[12] Although many people with ALS fear choking to death (suffocating),[19] they can be reassured that this occurs rarely, about 0–3% of the time. About 90% of people with ALS die peacefully.[119] In the final days of life, opioids can be used to treat pain and nafas qisilishi, esa benzodiazepinlar can be used to treat anxiety.[18]
Epidemiologiya
ALS is the most common motor neuron disease in adults and the third most common neurodegenerative disease[26] keyin Altsgeymer kasalligi va Parkinson kasalligi.[120] Worldwide the number of people who develop ALS yearly is estimated to be 1.9 people per 100,000 per year, while the number of people who have ALS at any given time is estimated to be about 4.5 people per 100,000.[121] In Europe, the number of new cases a year is about 2.6 people per 100,000, while the number affected is 7–9 people per 100,000.[7] The lifetime risk of developing ALS is 1:350 for European men and 1:400 for European women. Men have a higher risk mainly because spinal-onset ALS is more common in men than women.[21] The number of those with ALS in the United States in 2015 was 5.2 people per 100,000, and was higher in whites, males, and people over 60 years old.[22] The number of new cases is about 0.8 people per 100,000 per year in east Asia and about 0.7 people per 100,000 per year in south Asia. About 80% of ALS epidemiology studies have been conducted in Europe and the United States, mostly in people of northern European descent.[12] There is not enough information to determine the rates of ALS in much of the world, including Africa, parts of Asia, India, Russia, and South America.[21] There are several geographic clusters in the Western Pacific where the prevalence of ALS was reported to be 50–100 times higher than the rest of the world, including Guam, the Kii yarim oroli Yaponiya va G'arbiy Yangi Gvineya. The incidence in these areas has decreased since the 1960s;[1] the cause remains unknown.[21]
People of all races and ethnic backgrounds may be affected by ALS,[22] but it is more common in whites than in Africans, Asians, or Hispanics.[122] In the United States in 2015, the prevalence of ALS in whites was 5.4 people per 100,000, while the prevalence in blacks was 2.3 people per 100,000. The Midwest had the highest prevalence of the four US Census regions with 5.5 people per 100,000, followed by the Northeast (5.1), the South (4.7), and the West (4.4). The Midwest and Northeast likely had a higher prevalence of ALS because they have a higher proportion of whites than the South and West.[22] Ethnically mixed populations may be at a lower risk of developing ALS; a study in Cuba found that people of mixed ancestry were less likely to die from ALS than whites or blacks.[123] There are also differences in the genetics of ALS between different ethnic groups; the most common ALS gene in Europe is C9orf72, dan so'ng SOD1, TARDBPva FUS, while the most common ALS gene in Asia is SOD1, dan so'ng FUS, C9orf72va TARDBP.[124]

ALS can affect people at any age,[40] but the peak incidence is between 50–75 years[13] and decreases dramatically after 80 years.[3] The reason for the decreased incidence in the elderly is unclear. One thought is that people who survive into their 80s may not be genetically susceptible to developing ALS; alternatively, ALS in the elderly might go undiagnosed because of qo'shma kasalliklar (other diseases they have), difficulty seeing a neurologist, or dying quickly from an aggressive form of ALS.[123] In the United States in 2015, the lowest prevalence was in the 18–39 age group, while the highest prevalence was in the 70–79 age group.[22] Sporadic ALS usually starts around the ages of 58 to 63 years, while familial ALS starts earlier, usually around 47 to 52 years.[3] The number of ALS cases worldwide is projected to increase from 222,801 in 2015 to 376,674 in 2040, an increase of 69%. This will largely be due to the aging of the world's population, especially in developing countries.[122]
Tarix

Descriptions of the disease date back to at least 1824 by Charlz Bell.[23] 1850 yilda, Fransua-Amilkar Aran was the first to describe a disorder he named "progressive muscular atrophy", a form of ALS in which only the lower motor neurons are affected.[125] In 1869, the connection between the symptoms and the underlying neurological problems were first described by Jan-Martin Sharko, who initially introduced the term amiotrofik lateral skleroz in his 1874 paper.[23] Flail arm syndrome, a regional variant of ALS, was first described by Alfred Vulpian in 1886. Flail leg syndrome, another regional variant of ALS, was first described by Pierre Marie and his student Patrikios in 1918.[126]
In 1945, American naval doctors reported that ALS was 100 times more prevalent among the Chamorro xalqi ning Guam than in the rest of the world. In 1956 the variant of ALS endemic to Guam was named "amyotrophic lateral sclerosis/parkinsonism dementia complex" (ALS/PDC), as it had the typical symptoms of ALS accompanied by parkinsonizm -like symptoms; the name in the local language is lytico-bodig disease. Despite a number of genetic and environmental studies, the cause of ALS/PDC remains unknown. Rates peaked in the early 1950s and steadily declined thereafter, and by 1985 the incidence of ALS/PDC in Guam was about the same as the rest of the world.[127]
The first gene to be associated with ALS was SOD1, which was identified in 1993.[26] This led to the development of the first hayvon modeli of ALS, the transgenik SOD1 mouse, in 1994.[27] In December 1995, riluzole became the first FDA-approved drug for ALS. It was then approved in Europe in 1996 and in Japan in 1998.[102] In 1996, the ALS Functional Rating Scale (ALSFRS) was first published; it was a 10-item questionnaire that measured the ability of people with ALS to perform kundalik hayot faoliyati.[128] In 1999, the scale was changed to give more weight to respiratory symptoms. Natijada ALS Functional Rating Scale - Revised (ALSFRS-R) is a 12-item questionnaire that replaces the single question about breathing with a question each about dyspnea, orthopnea, and respiratory insufficiency.[129]
In 2006, it was discovered that the protein TDP-43 is a major component of the inclusion bodies seen in both ALS and frontotemporal dementia (FTD), which provided evidence that ALS and FTD are part of a common disease spectrum. This led to the discovery in 2008 that mutations in TARDBP, the gene that codes for TDP-43, are a cause of familial ALS.[26] In 2011, noncoding repeat expansions in C9orf72 were found to be a major cause of ALS and FTD.[12] Edaravone was approved to treat ALS in Japan and South Korea in 2015 and in the United States in 2017.[115] 2017 yildan boshlab[yangilash], it has not been approved to treat ALS in Europe.[114]
Diagnostika mezonlari
1950-yillarda, electrodiagnostic testing (EMG and NCV) began to be used to evaluate clinically suspected ALS. 1969 yilda Edward H. Lambert published the first EMG/NCS diagnostic criteria for ALS, consisting of four findings he considered to strongly support the diagnosis.[130] 1990 yilda Butunjahon nevrologiya federatsiyasi (WFN) held a meeting at El eskaliy, Spain, to come up with precise diagnostic criteria for ALS to help standardize clinical trials; the resulting "El Escorial" criteria were published in 1994.[131] In 1998, the WFN held another meeting to revise the criteria at Airlie House in Uorrenton, Virjiniya; the resulting "Airlie House" or "El Escorial Revised" criteria were published in 2000.[132] In 2006, a meeting was held on Avaji oroli in Japan to discuss how to use EMG and NCV tests to help diagnose ALS earlier; the resulting "Awaji" criteria were published in 2008.[90]
Ism

Other names for ALS include Charcot's disease, Lou Gehrig's disease, and motor neurone disease.[1] Amyotrophic dan keladi Yunoncha so'z amyotrophia: a- means "no", myo refers to "muscle", and trophia means "nourishment". Shuning uchun, amyotrophia means "no muscle nourishment,"[134] which describes the loss of signals motor neurons usually send to muscle cells;[135] this leads to the characteristic muscle atrofiya seen in people with ALS. Yanal identifies the areas in a person's spinal cord where the affected motor neurons that control muscle are located. Sclerosis means "scarring" or "hardening" and refers to the death of the motor neurons in the spinal cord.[134]
ALS is sometimes referred to as "Charcot's disease" because Jean-Martin Charcot was the first to connect the clinical symptoms with the pathology seen at autopsy. The term is ambiguous and can also refer to Charcot-Mari-Tish kasalligi va Charcot joint disease.[136] The British neurologist Russell Brain coined the term "motor neurone disease" in 1933 to reflect his belief that ALS, progressive bulbar palsy, and progressive muscular atrophy were all different forms of the same disease,[137] although "neurone" should be spelt "neuron".[138] In some countries, especially the United States, ALS is called "Lou Gehrig's disease",[133] after American baseball player Lou Gerig, who developed ALS in 1938, had to stop playing baseball in 1939, and died from it in 1941.[139]
In the United States and continental Europe, the terms "ALS" or "Lou Gehrig's disease" refer to all forms of the disease, including classical ALS, progressive bulbar palsy, progressive muscular atrophy, and primary lateral sclerosis.[140][36] In the United Kingdom and Australia, the term "motor neurone disease" is the name used for ALS; and other diseases that affect the motor neurons are separately treated motor neuron diseases.[141][140]
Jamiyat va madaniyat

In August 2014, a challenge went virusli online, commonly known as the "ALS Ice Bucket Challenge ".[142] Contestants fill a bucket full of ice and water, then state who nominated them to do the challenge, and nominate three other individuals of their choice to take part in it. The contestants then dump the buckets of ice and water onto themselves. However, it can be done in a different order. The contestants then donate at least AQSH$ 10 (or a similar amount in their local currency) to ALS research at the ALS assotsiatsiyasi, ALS terapiyasini rivojlantirish instituti, Kanadaning ALS Jamiyati yoki Motor neyron kasalliklari assotsiatsiyasi Buyuk Britaniyada. Any contestants who refuse to have the ice and water dumped on them are expected to donate at least US$100 to ALS research. 2015 yil iyul holatiga ko'ra[yangilash], the Ice Bucket Challenge had raised $115 million for the ALS Association.[143] Many celebrities have taken part in the challenge.[144] The Ice Bucket Challenge was credited with helping to raise funds that contributed to the discovery that the gene NEK1 may potentially contribute to the development for ALS.[145][146]
Tadqiqot
Model organizmlar

Many different organisms are used as models for studying ALS, including Saccharomyces cerevisiae (a species of yeast),[87] Caenorhabditis elegans (a roundworm), Drosophila melanogaster (the common fruit fly), Danio rerio (the zebrafish), Muskul mushak (the house mouse), and Rattus norvegicus (the common rat).[13] None of these models perfectly represents ALS in humans, partly because most animal models are based on gene overexpression, meaning that multiple copies of the mutant human gene are inserted into the transgenic model, and partly because the human nervous system is very different from that of other animals.[12]
The first animal model for ALS was the SOD1G93A transgenic mouse,[g] which was developed in 1994. It expresses about 20–24 copies of the mutant human SOD1 gen[147] and reproduces most of the clinical and pathological findings seen in ALS.[148] Although there are now over 20 different SOD1 mouse models, the SOD1G93A model remains both the most widely used SOD1 mouse model[147] and the most widely used ALS mouse model overall.[27] Much of the present understanding of ALS pathophysiology came from studying mouse models that overexpress mutant SOD1,[147] ayniqsa SOD1G93A sichqonlar.[27] However, many drug targets that were shown to be effective in the SOD1G93A transgenic mouse failed in clinical trials in humans; boshqa SOD1 models have had similar problems.[147] Most of these drugs were identified as potentially effective based on a single study in a rodent SOD1 model and then failed in clinical trials in patients who primarily had sporadic ALS.[87] It is thought that these clinical trials failed because SOD1 mutations account for only 2% of all ALS cases[147] and because the pathology of SOD1 ALS is thought to be distinct from all other types of ALS; it lacks the abnormal aggregations of TDP-43 protein or FUS protein seen in nearly all other cases of ALS.[26]
As of 2018, there are about 20 TARDBP mouse models, a dozen FUS mouse models, and a number of C9orf72, PFN1va UBQLN2 mouse models. There are also new methods of developing animal models, including viral transgenez, in which viruses are used to deliver mutant genes to an animal model, and CRISPR / Cas9, which can be used to give an animal model multiple mutated genes. Both of these methods are faster and cheaper than traditional methods of genetically engineering mice; they also allow scientists to study the effects of a mutation in mice of different genetic backgrounds, which better represents the genetic diversity seen in humans.[27]
Cellular models used to study ALS include the yeast Saccharomyces cerevisiae and rat or mouse motor neurons in culture. Small-animal models include the fruit fly, the roundworm C. elegans, and the zebrafish. Of the three, the fruit fly is the most widely used; it has a rapid life-cycle, short lifespan, a sophisticated nervous system, and many genetic tools available. C. elegans has a short life-cycle, is easy to manipulate genetically, and has a simple but well-understood nervous system. The zebrafish has transparent embryos that can be injected with DNA or RNA and has a lifespan of up to two years.[87] Induktsiyalangan pluripotent ildiz hujayralari (iPSCs) can be used to convert skin fibroblastlar into motor neurons.[13] It is now possible to generate iPSCs from people with ALS, which can then be converted into spinal motor neurons, which are useful for studying disease mechanisms and for testing potential drugs for ALS. iPSCs allow sporadic ALS to be modeled, which cannot be done with animal models.[87]
Muolajalar
From the 1960s until 2014, about 50 drugs for ALS were tested in randomized controlled trials (RCTs);[h] of these, riluzole was the only one that showed a slight benefit in improving survival. Drugs tested and not shown to be effective in clinical trials in humans include antiviral drugs, anti-excitotoxic drugs, growth factors, neurotrophic factors, anti-inflammatory drugs, antioxidants, anti-apoptotic drugs, and drugs to improve mitochondria function.[149]
An analysis of 23 large phase II and phase III RCTs that failed between 2004 and 2014 concluded that there were many potential reasons for their lack of success. These trials in humans went ahead on the basis of positive results in SOD1 transgenic mice, which are not a good animal model for sporadic ALS. Additionally, in most preclinical studies the SOD1 mice were given the drug during the presymptomatic stage; this makes the results less likely to apply to people with ALS, who begin treatment well after their symptoms begin. Positive results in small phase II studies in humans could also be misleading and lead to failure in phase III trials. Other potential issues included the drug not reaching its intended site of action in the central nervous system and dorilarning o'zaro ta'siri between the study drug and riluzole.[149]
Takroriy transkranial magnit stimulyatsiya had been studied in ALS in small and poorly designed clinical trials; 2013 yildan boshlab[yangilash], evidence was insufficient to know whether rTMS is safe or effective for ALS.[150] One 2016 review of ildiz hujayralari terapiyasi trials found tentative evidence that intraspinal stem cell implantation was relatively safe and possibly effective.[151] 2019 yil Cochrane-ni ko'rib chiqish of cell-based therapies found that there was insufficient evidence to speculate about efficacy.[152] Masitinib has been approved as an yetim dori in Europe and the United States, with studies ongoing as of 2016[yangilash].[153] Beta-adrenergik agonist drugs have been proposed as a treatment for their effects on muscle growth and neuroprotection, but research in humans is insufficient to determine their efficacy.[154]
Sababi
With the discovery that TDP-43, FUS va C9orf72 can cause ALS as well as related forms of frontotemporal dementia (FTD/ALS)[155][156] there has been intense effort to understand how these mutations cause disease, and whether other protein dysfunction may be important. 2013 yildan boshlab[yangilash] it appeared that differences in the metilatsiya of arginine residues in FUS protein may be relevant, and methylation status may be a way to distinguish some forms of FTD from ALS.[157]
Shuningdek qarang
Izohlar
- ^ Additional names for flail arm syndrome include the scapulohumeral form of ALS, Vulpian–Bernart syndrome, hanging arm syndrome, and neurogenic man-in-a-barrel syndrome.[20]
- ^ Additional names for flail leg syndrome that involves both lower legs (bilateral distal involvement) include pseudopolyneuritic ALS, Patrikios syndrome, Marie-Patrikios ALS, and the peroneal form of ALS.[20]
- ^ According to one cohort study, 11.5% of people with ALS have extraocular muscle dysfunction.[38]
- ^ In 2013, the NFL reached a $765 million agreement to compensate more than five thousand former NFL players for concussion-related injuries and illnesses.[78] Some NFL players involved in the legal settlement complained that the NFL was not doing enough to help players. The judge in the case concurred, and in 2015 the NFL agreed to pay an unlimited amount of damages for players found to have ALS, Parkinson kasalligi, Altsgeymer kasalligi, or dementia.[79]
- ^ The criteria are "scores of at least 2 points on all 12 items of ALSFRS-R, forced vital capacity of 80% or more, definite or probable ALS according to the revised El Escorial criteria, and disease duration of 2 years or less."[101]
- ^ Based on population-based ALS registries, it is estimated that less than 7% of people with ALS meet these criteria.[113]
- ^ "G93A" means that the 93rd amino acid residue in the SOD1 protein has been changed from glycine to alanine.
- ^ To'liq ro'yxat uchun qarang Amyotrophic lateral sclerosis research#Past clinical trials.
Adabiyotlar
- ^ a b v Wijesekera LC, Leigh PN (February 2009). "Amiotrofik lateral skleroz". Noyob kasalliklar jurnali. 3 (4): 3. doi:10.1186/1750-1172-4-3. PMC 2656493. PMID 19192301.
- ^ a b v d e f g h men j k l m n o p q r s t siz v w x y "Amyotrophic Lateral Sclerosis (ALS) Fact Sheet | National Institute of Neurological Disorders and Stroke". www.ninds.nih.gov. Olingan 22 oktyabr 2020.
- ^ a b v d e f g h men j k Kiernan MC, Vucic S, Cheah BC, Turner MR, Eisen A, Hardiman O, Burrell JR, Zoing MC (March 2011). "Amiotrofik lateral skleroz". Lanset. 377 (9769): 942–55. doi:10.1016/s0140-6736(10)61156-7. PMID 21296405.
- ^ a b v d e f g h men j k l m n o p q r s t siz v w Hobson EV, McDermott CJ (September 2016). "Amiotrofik lateral sklerozni qo'llab-quvvatlovchi va simptomatik boshqarish" (PDF). Tabiat sharhlari. Nevrologiya. 12 (9): 526–38. doi:10.1038 / nrneurol.2016.111. PMID 27514291. S2CID 8547381.
- ^ a b v d e f g Miller RG, Mitchell JD, Mur DH (mart 2012). "Amilotrofik lateral skleroz (ALS) / motorli neyron kasalligi (MND) uchun riluzol". Tizimli sharhlarning Cochrane ma'lumotlar bazasi. 3 (3): CD001447. doi:10.1002 / 14651858.CD001447.pub3. PMC 7055506. PMID 22419278.
- ^ "FDA ALSni davolash uchun dori-darmonlarni tasdiqlaydi". AQSh oziq-ovqat va farmatsevtika idorasi. 2017 yil 5-may. Arxivlandi asl nusxasidan 2017 yil 8 mayda.
- ^ a b v Hardiman O, Al-Chalabi A, Brayne C, Beghi E, van den Berg LH, Chio A, Martin S, Logroscino G, Runi J (iyul 2017). "Amiotrofik lateral sklerozning o'zgaruvchan surati: Evropa registrlaridan darslar". Nevrologiya, neyroxirurgiya va psixiatriya jurnali. 88 (7): 557–63. doi:10.1136 / jnnp-2016-314495. PMID 28285264. S2CID 52871105.
- ^ a b v d e "Motor neyron kasalligi - NHS". nhs.uk. 2018 yil 15-yanvar. Olingan 24 oktyabr 2020.
- ^ Avstraliya, Healthdirect (2020 yil 17 aprel). "Dvigatel neyron kasalligi (MND)". www.healthdirect.gov.au. Olingan 24 oktyabr 2020.
- ^ Zucchi E, Bonetto V, Sorarù G va boshq. (15 oktyabr 2020). "Motor neyron kasalliklarida neyrofilamentlar: istiqbolli diagnostika va prognostik biomarkerlar tomon". Molekulyar neyrodejeneratsiya. 15 (1): 58. doi:10.1186 / s13024-020-00406-3. PMID 33059698. S2CID 222385359.
- ^ "Dvigatel neyron kasalliklari to'g'risida ma'lumot varaqasi | Milliy nevrologik kasalliklar va qon tomir instituti". www.ninds.nih.gov. Olingan 27 oktyabr 2020.
- ^ a b v d e f g h men j k l m n o p q r s t siz v w x y z aa Hardiman O, Al-Chalabi A, Chio A, Corr EM, Logroscino G, Robberecht V, Shaw PJ, Simmons Z, van den Berg LH (oktyabr 2017). "Amiotrofik lateral skleroz" (PDF). Tabiat sharhlari. Kasalliklarga qarshi vositalar. 3 (17071): 17071. doi:10.1038 / nrdp.2017.71. PMID 28980624. S2CID 1002680.
- ^ a b v d e f g h men j k l m n o p q r s t siz van Es MA, Hardiman O, Chio A, Al-Chalabi A, Pasterkamp RJ, Veldink JH, van den Berg LH (2017 yil noyabr). "Amiotrofik lateral skleroz". Lanset. 390 (10107): 2084–2098. doi:10.1016 / S0140-6736 (17) 31287-4. PMID 28552366. S2CID 24483077.
- ^ a b v Chiò A, Mora G, Lauria G (fevral 2017). "Amiotrofik lateral sklerozdagi og'riq". Lanset. Nevrologiya. 16 (2): 144–57. arXiv:1607.02870. doi:10.1016 / S1474-4422 (16) 30358-1. PMID 27964824. S2CID 38905437.
- ^ Xilton JB, Oq AR, Crouch PJ (may, 2015). "Amiotrofik lateral sklerozda metall tanqisligi SOD1". Molekulyar tibbiyot jurnali (Berlin, Germaniya). 93 (5): 481–7. doi:10.1007 / s00109-015-1273-3. PMID 25754173. S2CID 12043749.
- ^ a b "ALS haqida tushuncha". ALS assotsiatsiyasi.
- ^ a b v Wingo TS, Cutler DJ, Yarab N, Kelly CM, Glass JD (2011). "Amerika Qo'shma Shtatlarining klinik jihatdan aniqlangan tadqiqot registrida amiotrofik lateral sklerozning merosxo'rligi". PLOS ONE. 6 (11): e27985. Bibcode:2011PLoSO ... 627985W. doi:10.1371 / journal.pone.0027985. PMC 3222666. PMID 22132186.
- ^ a b v d e f g h men j k l m n Soriani M, Desnuelle C (2017 yil may). "Amiotrofik lateral sklerozda yordamni boshqarish". Revue Neurologique. 173 (5): 288–89. doi:10.1016 / j.neurol.2017.03.031. PMID 28461024.
- ^ a b v d e f g h men j k Connolly S, Galvin M, Hardiman O (aprel 2015). "Amiotrofik lateral skleroz bilan og'rigan bemorlarda umrining oxirini boshqarish". Lanset. Nevrologiya. 14 (4): 435–42. doi:10.1016 / S1474-4422 (14) 70221-2. PMID 25728958. S2CID 34109901.
- ^ a b v d e f g h Swinnen B, Robberecht V (noyabr 2014). "Amiotrofik lateral sklerozning fenotipik o'zgaruvchanligi". Tabiat sharhlari. Nevrologiya. 10 (11): 661–70. doi:10.1038 / nrneurol.2014.184. PMID 25311585. S2CID 205516010.
- ^ a b v d e f g Al-Chalabi A, Hardiman O (2013 yil noyabr). "ALS epidemiologiyasi: genlar, atrof-muhit va vaqtning fitnasi". Tabiat sharhlari. Nevrologiya. 9 (11): 617–28. doi:10.1038 / nrneurol.2013.203 yil. PMID 24126629. S2CID 25040863.
- ^ a b v d e f Mehta P, Kaye V, Raymond J, Panjabi R, Larson T, Koen J, Muravov O, Xorton K (2018 yil noyabr). "Amiotrofik lateral sklerozning tarqalishi - Amerika Qo'shma Shtatlari, 2015 yil". Kasallik va o'lim bo'yicha haftalik hisobot. 67 (46): 1285–1289. doi:10.15585 / mmwr.mm6746a1. PMC 5858037. PMID 30462626.
- ^ a b v d e Rowland LP (2001 yil mart). "Qanday qilib amiotrofik lateral skleroz o'z nomini oldi: Jan-Martin Sharkoning klinik-patologik dahosi". Nevrologiya arxivi. 58 (3): 512–15. doi:10.1001 / archneur.58.3.512. PMID 11255459.
- ^ Kelly, Evelyn B. (2013). Inson genetikasi va kasalliklari entsiklopediyasi. Santa Barbara, Kaliforniya: Grinvud. 79-80 betlar. ISBN 978-0-313-38713-5. Arxivlandi asl nusxasidan 2017 yil 8 sentyabrda.
- ^ Youngson, David B. Jacoby, Robert M. (2004). Oila salomatligi ensiklopediyasi (3-nashr). Tarritaun, Nyu-York: Marshall Kavendish. p. 1256. ISBN 978-0-7614-7486-9. Arxivlandi asl nusxasidan 2017 yil 8 sentyabrda.
- ^ a b v d e f g h Renton AE, Chiò A, Traynor BJ (yanvar 2014). "Amiotrofik lateral skleroz genetikasidagi o'yin holati". Tabiat nevrologiyasi. 17 (1): 17–23. doi:10.1038 / nn.3584. hdl:2318/156177. PMC 4544832. PMID 24369373.
- ^ a b v d e Lutz C (2018 yil avgust). "ALS-ning sichqoncha modellari: o'tmishi, hozirgi va kelajak". Miya tadqiqotlari. 1693 (A qism): 1-10. doi:10.1016 / j.brainres.2018.03.024. PMID 29577886. S2CID 4641251.
- ^ Qo'shiq P (2014 yil avgust). "Ice Bucket Challenge: davlat sektori kamdan kam uchraydigan kasalliklarga tibbiy yordam ko'rsatish va izlanishlar tizimini barqaror rivojlanishiga zudlik bilan yordam berishga tayyor bo'lishi kerak". Murakkab va noyob kasalliklarni tadqiq qilish. 3 (3): 94–96. doi:10.5582 / irdr.2014.01015. PMC 4214244. PMID 25364651.
- ^ "8B60 Motor neyron kasalligi". O'lim va kasallanish statistikasi bo'yicha ICD-11. Jahon Sog'liqni saqlash tashkiloti. Olingan 24 yanvar 2019.
- ^ a b v Jawdat O, Statland JM, Barohn RJ, Katz JS, Dimachkie MM (noyabr 2015). "Amiotrofik lateral sklerozning mintaqaviy variantlari (brakiyal amiotrofik diplegiya, oyoq amyotrofik diplegiya va izolyatsiyalangan bulbar amyotrofik lateral skleroz)". Nevrologik klinikalar. 33 (4): 775–85. doi:10.1016 / j.ncl.2015.07.003. PMC 4629514. PMID 26515621.
- ^ a b v d e f Grad LI, Rouleau GA, Ravits J, Cashman NR (avgust 2017). "Amiotrofik lateral sklerozning klinik spektri (ALS)". Tibbiyotda sovuq bahor porti istiqbollari. 7 (8): a024117. doi:10.1101 / cshperspect.a024117. PMC 5538408. PMID 28003278.
- ^ a b v d Chiò A, Calvo A, Moglia C, Mazzini L, Mora G (iyul 2011). "Amiotrofik lateral sklerozning fenotipik heterojenligi: populyatsiyani o'rganish". Nevrologiya, neyroxirurgiya va psixiatriya jurnali. 82 (7): 740–46. doi:10.1136 / jnnp.2010.235952. PMID 21402743. S2CID 13416164.
- ^ Gautier G, Verschueren A, Monnier A, Attorian S, Salort-Campana E, Puget J (avgust 2010). "Nafas olish boshlangan ALS: Klinik xususiyatlari va invaziv bo'lmagan shamollatishning prognozga ta'siri". Amiotrofik lateral skleroz. 11 (4): 379–82. doi:10.3109/17482960903426543. PMID 20001486. S2CID 27672209.
- ^ a b v d e Al-Chalabi A, Hardiman O, Kiernan MC, Chiò A, Rix-Brooks B, van den Berg LH (oktyabr 2016). "Amiotrofik lateral skleroz: yangi tasniflash tizimiga o'tish". Lanset. Nevrologiya. 15 (11): 1182–94. doi:10.1016 / S1474-4422 (16) 30199-5. hdl:2318/1636249. PMID 27647646. S2CID 45285510.
- ^ Teoh HL, Carey K, Sampaio H, Mowat D, Roscioli T, Farrar M (2017). "Pediatrik motorli neyron kasalliklari: o'murtqa mushak atrofiyasidan tashqari". Asab plastisiyasi. 2017: 6509493. doi:10.1155/2017/6509493. PMC 5467325. PMID 28634552.
- ^ a b Tard C, Defebvre L, Mau S, Devos D, Danel-Brunaud V (may 2017). "Amiotrofik lateral sklerozning klinik xususiyatlari va ularning prognostik qiymati". Revue Neurologique. 173 (5): 263–72. doi:10.1016 / j.neurol.2017.03.029. PMID 28477850.
- ^ Lui AJ, Byl NN (iyun 2009). "O'rtacha intensiv mashqlar funktsiyasi va amiotrofik lateral sklerozda kasallikning rivojlanishiga ta'sirini tizimli ko'rib chiqish". Nevrologik jismoniy terapiya jurnali. 33 (2): 68–87. doi:10.1097 / NPT.0b013e31819912d0. PMID 19556916. S2CID 7650356.
- ^ McCluskey L, Vandriel S, Elman L, Van Deerlin VM, Powers J, Boller A va boshq. (2014 yil 15 oktyabr). "ALS-Plus sindromi: katta ALS kohortasidagi piramidal bo'lmagan xususiyatlar". Nevrologiya fanlari jurnali. 345 (1–2): 118–24. doi:10.1016 / j.jns.2014.07.022. PMC 4177937. PMID 25086858.
- ^ a b v d e f g h men j Jigarrang RH, Al-Chalabi A (13 iyul 2017). "Amiotrofik lateral skleroz". Nyu-England tibbiyot jurnali. 377 (2): 162–172. doi:10.1056 / NEJMra1603471. PMID 28700839.
- ^ a b v Martin S, Al Khleifat A, Al-Chalabi A (2017). "Amiotrofik lateral skleroz nima sabab bo'ladi?". F1000Qidiruv. 6: 371. doi:10.12688 / f1000research.10476.1. PMC 5373425. PMID 28408982.
- ^ Raaphorst J, Beeldman E, De Visser M, De Haan RJ, Shmand B (oktyabr 2012). "Motor neyronlari kasalliklarida xatti-harakatlarning o'zgarishini muntazam ravishda ko'rib chiqish". Amiotrofik lateral skleroz. 13 (6): 493–501. doi:10.3109/17482968.2012.656652. PMID 22424127. S2CID 22224140.
- ^ a b Beeldman E, Raaphorst J, Klein Twennaar M, de Visser M, Schmand BA, de Haan RJ (iyun 2016). "ALSning kognitiv profili: muntazam tahlil va meta-tahlilni yangilash". Nevrologiya, neyroxirurgiya va psixiatriya jurnali. 87 (6): 611–19. doi:10.1136 / jnnp-2015-310734. PMID 26283685. S2CID 22082109.
- ^ Gordon PH, Miller RG, Mur DH (sentyabr 2004). "ALSFRS-R". Amiotrofik lateral skleroz va boshqa vosita neyron kasalliklari. 5 Qo'shimcha 1: 90-93. doi:10.1080/17434470410019906. PMID 15512883. S2CID 218987659.
- ^ Creemers H, Grupstra H, Nollet F, van den Berg LH, Beelen A (iyun 2015). "ALS bilan kasallangan bemorlarning funktsional holati uchun prognostik omillar: tizimli tekshiruv". Nevrologiya jurnali. 262 (6): 1407–23. doi:10.1007 / s00415-014-7564-8. PMID 25385051. S2CID 31734765.
- ^ Castrillo-Viguera C, Grasso DL, Simpson E, Shefner J, Cudkowicz ME (2010). "ALSFRS-R pasayishi o'zgarishidagi klinik ahamiyat". Amiotrofik lateral skleroz (Jurnal maqolasi). 11 (1–2): 178–80. doi:10.3109/17482960903093710. PMID 19634063. S2CID 207619689.
- ^ Sabatelli M, Madia F, Conte A, Luigetti M, Zollino M, Mancuso I, Lo Monako M, Lippi G, Tonali P (sentyabr 2008). "Yosh amyotrofik lateral sklerozning tabiiy tarixi". Nevrologiya. 71 (12): 876–81. doi:10.1212 / 01.wnl.0000312378.94737.45. PMID 18596241. S2CID 10848454.
- ^ Paganoni S, Deng J, Yaffa M, Cudkovich ME, Wills AM (iyul 2011). "Dislipidemiya emas, tana massasi indeksi amyotrofik lateral sklerozda omon qolish uchun mustaqil bashoratchi hisoblanadi". Mushak va asab. 44 (1): 20–24. doi:10.1002 / mus.22114. PMC 4441750. PMID 21607987. Xulosa – Massachusets umumiy kasalxonasi (2011 yil 11-may).
- ^ "Stiven Xoking ALS kasallari uchun namuna bo'lib xizmat qiladi". CNN. 2009 yil 20 aprel. Arxivlandi asl nusxasidan 2016 yil 15 avgustda.
- ^ "Familial ALS - ALS tadqiqotlari bo'yicha hamkorlik". Olingan 27 oktyabr 2020.
- ^ a b Vajda A, McLaughlin RL, Heverin M, Thorpe O, Abrahams S, Al-Chalabi A, Hardiman O (mart 2017). "ALSdagi genetik sinov: amaldagi amaliyotlarni o'rganish". Nevrologiya. 88 (10): 991–999. doi:10.1212 / WNL.0000000000003686. PMC 5333513. PMID 28159885.
- ^ Byrne S, Walsh C, Lynch C, Bede P, Elamin M, Kenna K, McLaughlin R, Hardiman O (iyun 2011). "Oilaviy amiotrofik lateral skleroz darajasi: sistematik tahlil va meta-tahlil". Nevrologiya, neyroxirurgiya va psixiatriya jurnali. 82 (6): 623–7. doi:10.1136 / jnnp.2010.224501. hdl:2262/53330. PMID 21047878. S2CID 6254190.
- ^ a b v U J, Mangelsdorf M, Fan D, Bartlett P, Braun MA (dekabr 2015). "Amiotrofik lateral skleroz genetik tadqiqotlar: Genom bo'yicha assotsiatsiyani xaritalashdan genom ketma-ketligiga" (PDF). Nevrolog. 21 (6): 599–615. doi:10.1177/1073858414555404. PMID 25378359. S2CID 3437565.
- ^ a b Corcia P, Couratier P, Blasco H, Andres CR, Beltran S, Meininger V, Vourch P (may 2017). "Amiotrofik lateral sklerozning genetikasi". Revue Neurologique. 173 (5): 254–262. doi:10.1016 / j.neurol.2017.03.030. PMID 28449881.
- ^ Chia R, Chiò A, Traynor BJ (yanvar 2018). "Amiotrofik lateral skleroz bilan bog'liq yangi genlar: diagnostikasi va klinik ta'siri". Lanset. Nevrologiya. 17 (1): 94–102. doi:10.1016 / S1474-4422 (17) 30401-5. PMC 5901717. PMID 29154141.
- ^ Zou ZY, Liu CY, Che CH, Huang HP (yanvar 2016). "Amiotrofik lateral sklerozda aniq tibbiyotga qarab". Translational Medicine yilnomalari. 4 (2): 27. doi:10.3978 / j.issn.2305-5839.2016.01.16. PMC 4731596. PMID 26889480.
- ^ Sontxaymer, Xarald (2015). Asab tizimining kasalliklari. Akademik matbuot. p. 170. ISBN 978-0-12-800403-6. Arxivlandi asl nusxasidan 2017 yil 8 sentyabrda. Olingan 2 may 2015.
- ^ Couratier P, Corcia P, Lautrette G, Nicol M, Marin B (may 2017). "ALS va frontotemporal demans umumiy kasallik spektriga tegishli". Revue Neurologique. 173 (5): 273–279. doi:10.1016 / j.neurol.2017.04.001. PMID 28449882.
- ^ a b Nguyen HP, Van Broekxoven S, van der Zee J (iyun 2018). "Genomik davrdagi ALS genlari va ularning FTDga ta'siri". Genetika tendentsiyalari. 34 (6): 404–423. doi:10.1016 / j.tig.2018.03.001. PMID 29605155.
- ^ a b Soqol JD, Kamel F (1 yanvar 2015). "Amiotrofik skleroz etiologiyasi va omon qolish bilan bog'liq harbiy xizmat, joylashuvlar va ta'sirlar". Epidemiologik sharhlar. 37 (1): 55–70. doi:10.1093 / epirev / mxu001. PMC 4325667. PMID 25365170.
- ^ Belbasis L, Bellou V, Evangelou E (mart 2016). "Atrof-muhit xavfi omillari va amiotrofik lateral skleroz: soyabonni ko'rib chiqish va kuzatuv tadqiqotlarining tizimli sharhlari va meta-tahlillaridan hozirgi dalillarni tanqidiy baholash". Neyroepidemiologiya. 46 (2): 96–105. doi:10.1159/000443146. PMID 26731747. S2CID 13163292.
- ^ Soqol JD, Steege AL, Ju J, Lu J, Luckhaupt SE, Schubauer-Berigan MK (iyul 2017). "Turli xil kasb guruhlari orasida amiotrofik lateral skleroz va Parkinson kasalligi o'limi - Amerika Qo'shma Shtatlari, 1985-2011". MMWR. Kasallik va o'lim bo'yicha haftalik hisobot. 66 (27): 718–722. doi:10.15585 / mmwr.mm6627a2. PMC 5687590. PMID 28704346.
- ^ Suteja NA, Fischer K, Veldink JH, van der Heijden GJ, Kromhout H, Heederik D, Huisman MH, Wokke JJ, van den Berg LH (2009). "Biz ALS uchun xavfli omil sifatida ishg'ol haqida haqiqatan ham bilamiz: tanqidiy va tizimli ko'rib chiqish". Amiotrofik lateral skleroz. 10 (5–6): 295–301. doi:10.3109/17482960802430799. PMID 19922116. S2CID 25772664.
- ^ Ingre C, Roos PM, Piehl F, Kamel F, Fang F (2015). "Amiotrofik lateral skleroz uchun xavf omillari". Klinik epidemiologiya. 7: 181–93. doi:10.2147 / CLEP.S37505. PMC 4334292. PMID 25709501.
- ^ Kamel F, Umbach DM, Bedlack RS, Richards M, Watson M, Alavanja MC, Bler A, Hoppin JA, Shmidt S, Sandler DP (iyun 2012). "Pestitsidlarga ta'sir qilish va amiotrofik lateral skleroz". Neyrotoksikologiya. 33 (3): 457–62. doi:10.1016 / j.neuro.2012.04.001. PMC 3358481. PMID 22521219.
- ^ Bozzoni V, Pansarasa O, Diamanti L, Nosari G, Cereda C, Ceroni M (2016). "Amiotrofik lateral skleroz va atrof-muhit omillari". Funktsional nevrologiya. 31 (1): 7–19. PMC 4819821. PMID 27027889.
- ^ Malek AM, Barchovskiy A, Bowser R, Youk A, Talbott EO (avgust 2012). "Pestitsid ta'sir qilish amiotrofik lateral skleroz uchun xavf omili sifatida: epidemiologik tadqiqotlar metanahlil: pestitsid ta'sir qilish ALS uchun xavf omili sifatida". Atrof-muhit tadqiqotlari. 117: 112–9. Bibcode:2012ER .... 117..112M. doi:10.1016 / j.envres.2012.06.007. PMID 22819005.
- ^ a b Gardner RC, Yaffe K (may, 2015). "Yengil shikastlanadigan miya shikastlanishi va neyrodejenerativ kasallik epidemiologiyasi". Molekulyar va hujayra nevrologiyalari. 66 (Pt B): 75-80. doi:10.1016 / j.mcn.2015.03.001. PMC 4461453. PMID 25748121.
- ^ Vatanabe Y, Vatanabe T (oktyabr 2017). "Bosh jarohati va amiotrofik lateral skleroz xavfi o'rtasidagi bog'liqlikni meta-analitik baholash". Evropa epidemiologiya jurnali. 32 (10): 867–879. doi:10.1007 / s10654-017-0327-y. PMID 29080013. S2CID 449855.
- ^ Luna, J .; Logroscino, G.; Kurschi, P.; Marin, B. (2017 yil may). "ALS epidemiologiyasining dolzarb muammolari: populyatsiya va jismoniy faollik o'rtasida ALS paydo bo'lishining o'zgarishi xavf omilidir". Revue Neurologique. 173 (5): 244–253. doi:10.1016 / j.neurol.2017.03.035. ISSN 0035-3787. PMID 28477849.
- ^ Veldink JH, Kalmijn S, Groeneveld GJ, Titulaer MJ, Vokke JH, van den Berg LH (2005 yil yanvar). "Jismoniy faollik va sporadik ALS bilan bog'liqlik". Nevrologiya. 64 (2): 241–5. doi:10.1212 / 01.WNL.0000149513.82332.5C. PMID 15668420. S2CID 36449771.
- ^ Armon C (2007 yil noyabr). "Amiotrofik lateral sklerozda sport va travma qayta ko'rib chiqildi". Nevrologiya fanlari jurnali. 262 (1–2): 45–53. doi:10.1016 / j.jns.2007.06.021. PMID 17681549. S2CID 20733887.
- ^ a b Harwood KA, McDermott CJ, Shaw PJ (avgust 2009). "Jismoniy faollik motorli neyron kasalligining ekzogen xavf omili sifatida (MND): dalillarni qayta ko'rib chiqish". Amiotrofik lateral skleroz. 10 (4): 191–204. doi:10.1080/17482960802549739. PMID 19263258. S2CID 14749160.
- ^ Hamidou B, Couratier P, Besancon C, Nicol M, Preux PM, Marin B (iyul 2014). "Jismoniy faollik ALS uchun xavfli omil emasligini ko'rsatadigan epidemiologik dalillar". Evropa epidemiologiya jurnali. 29 (7): 459–75. doi:10.1007 / s10654-014-9923-2. PMID 24986107. S2CID 20563636.
- ^ Lacorte E, Ferrigno L, Leoncini E, Corbo M, Boccia S, Vanacore N (2016 yil iyul). "Sport, bo'sh vaqt va kasb faoliyati bilan bog'liq jismoniy faollik va jismoniy faollik ALS uchun xavf omillari sifatida: muntazam ravishda qayta ko'rib chiqish". Neyrologiya va biobehavioral sharhlar. 66: 61–79. doi:10.1016 / j.neubiorev.2016.04.007. PMID 27108217. S2CID 24844638.
- ^ Couratier P, Corcia P, Lautrette G, Nicol M, Preux P, Marin B (yanvar 2016). "Amiotrofik lateral skleroz epidemiologiyasi: Adabiyotga obzor". Neyroepidemiologiya. 172 (1): 37–45. doi:10.1016 / j.neurol.2015.11.002. PMID 26727307.
- ^ Tharmaratnam T, Iskandar M, Tabobondung T, Tobbia I, Gopee-Ramanan P, Tabobondung T (2018 yil 19-iyun). "Professional amerikalik futbol o'yinchilaridagi surunkali travmatik ensefalopatiya: biz hozir qayerdamiz?". Nevrologiyaning chegaralari. 19 (9): 445. doi:10.3389 / fneur.2018.00445. PMC 6018081. PMID 29971037.
- ^ Gardner A, Iverson G, Makkrori P (yanvar 2014). "Sportdagi surunkali travmatik ensefalopatiya: tizimli ko'rib chiqish". Britaniya sport tibbiyoti jurnali. 48 (2): 84–90. doi:10.1136 / bjsports-2013-092646. PMID 23803602. S2CID 7182895.
- ^ Belson, Ken (22 aprel 2015). "Sudya N.F.L. sarsıntı kostyumidagi kelishuvni ma'qulladi". The New York Times. Olingan 10 avgust 2018.
- ^ "Kevin Tyorner, keyinchalik ligada kurash olib borgan N.F.L. futbolchisi 46 yoshida vafot etdi". The New York Times. 2016 yil 24 mart. Arxivlandi asl nusxasidan 2016 yil 28 martda. Olingan 27 mart 2016.
- ^ Armon C (2009 yil noyabr). "Chekish vaqti-vaqti bilan paydo bo'lgan ALS uchun xavf omilidir.". Nevrologiya. 73 (20): 1693–8. doi:10.1212 / WNL.0b013e3181c1df48. PMC 2788806. PMID 19917993.
- ^ Alonso A, Logroscino G, Hernan MA (Noyabr 2010). "Chekish va amiotrofik lateral skleroz xavfi: sistematik tahlil va meta-tahlil". Nevrologiya, neyroxirurgiya va psixiatriya jurnali. 81 (11): 1249–52. doi:10.1136 / jnnp.2009.180232. PMID 20639382. S2CID 2079442.
- ^ Vang H, O'Reilly ÉJ, Vayskopf MG, Logroscino G, Makkullou ML, Thun MJ, Shatskin A, Kolonel LN, Ascherio A (Fevral 2011). "Chekish va amiotrofik lateral skleroz xavfi: 5 ta istiqbolli kohortlarning birlashtirilgan tahlili". Nevrologiya arxivi. 68 (2): 207–13. doi:10.1001 / archneurol.2010.367. PMC 3319086. PMID 21320987.
- ^ Robberecht V, Flibs T (2013 yil aprel). "Amiotrofik lateral sklerozning o'zgaruvchan sahnasi". Tabiat sharhlari. Nevrologiya. 14 (4): 248–64. doi:10.1038 / nrn3430. PMID 23463272. S2CID 208941.
- ^ Okamoto K, Mizuno Y (aprel 2008). "Aminotrofik lateral sklerozda Bunina tanalari". Neyropatologiya. 28 (2): 109–15. doi:10.1111 / j.1440-1789.2007.00873.x. PMID 18069968. S2CID 34398467.
- ^ Oq JA, Banerji R, Gunavardena S (2016 yil 19-may). "Akson transporti va neyrodejeneratsiya: dengiz dori-darmonlarini terapevtikani rivojlantirish uchun qanday ishlatish mumkin". Dengiz dori vositalari. 14 (5): 102. doi:10.3390 / md14050102. PMC 4882576. PMID 27213408.
- ^ Ng Kee Kwong, Koy Chong; Mehta, Arpan R.; Nedergaard, Mayken; Chandran, Siddxartan (2020 yil 20-avgust). "ALS patofiziologiyasida miya omurilik suyuqligining yangi funktsiyalarini aniqlash". Acta Neuropathologica Communications. 8 (1): 140. doi:10.1186 / s40478-020-01018-0. ISSN 2051-5960. PMC 7439665. PMID 32819425.
- ^ a b v d e Filipp Van Damm P, Robberex V, Van Den Bosch L (2017 yil may). "Amiotrofik lateral sklerozni modellashtirish: taraqqiyot va imkoniyatlar". Kasallik modellari va mexanizmlari. 10 (5): 537–49. doi:10.1242 / dmm.029058. PMC 5451175. PMID 28468939.
- ^ Xu Z, Xenderson RD, Devid M, Makkombe PA (2016). "Nörofilamentlar amiotrofik lateral sklerozning biomarkerlari sifatida: tizimli tahlil va meta-tahlil". PLOS ONE. 11 (10): e0164625. Bibcode:2016PLoSO..1164625X. doi:10.1371 / journal.pone.0164625. PMC 5061412. PMID 27732645.
- ^ Vu LT, Bowser R (2017 yil yanvar). "Amiotrofik lateral skleroz uchun suyuqlikka asoslangan biomarkerlar". Neyroterapevtikalar. 14 (1): 119–134. doi:10.1007 / s13311-016-0503-x. PMC 5233638. PMID 27933485.
- ^ a b de Carvalho M, Dengler R, Eisen A, Angliya JD, Kaji R, Kimura J va boshq. (2008 yil mart). "ALS diagnostikasining elektrodiagnostik mezonlari". Klinik neyrofiziologiya. 119 (3): 497–503. doi:10.1016 / j.clinph.2007.09.143. PMID 18164242. S2CID 14851649.
- ^ Kosta J, Svash M, de Karvalyu M (2012 yil noyabr). "Amiotrofik lateral skleroz diagnostikasi uchun avaji mezonlari: tizimli ko'rib chiqish". Nevrologiya arxivi. 69 (11): 1410–1416. doi:10.1001 / archneurol.2012.254. PMID 22892641.
- ^ Belsh JM (2000 yil mart). "El Escorial Revisited-ning ALS diagnostik mezonlari: ular klinisyenlarning, shuningdek tadqiqotchilarning ehtiyojlariga javob beradimi?". Amiotrofik lateral skleroz va boshqa vosita neyron kasalliklari. 1 (1-qo'shimcha): S57-S60. doi:10.1080/14660820052415925. PMID 11464928. S2CID 6828235.
- ^ Silani V, Messina S, Poletti B, Morelli C, Doretti A, Ticozzi N, Maderna L (mart 2011). "2010 yilda amiotrofik lateral skleroz diagnostikasi". Italiya de Biologie arxivi. 149 (1): 5–27. doi:10.4449 / aib.v149i1.1260. PMID 21412713.
- ^ "Lambert-Eaton Myasthenic Syndrome (LEMS)". Misc.medscape.com. Arxivlandi asl nusxasidan 2013 yil 14 mayda. Olingan 18 aprel 2013.
- ^ "LEMS.com, Lambert-Eaton Myasthenic Syndrome: About". Lems.com. Arxivlandi asl nusxasidan 2013 yil 20 yanvarda. Olingan 18 aprel 2013.
- ^ Mills KR (2010 yil noyabr). "Amiotrofik lateral skleroz va benign fasikulyatsiya sindromidagi fatsikulyatsiyalarning xususiyatlari". Miya. 133 (11): 3458–69. doi:10.1093 / brain / awq290. PMID 20959307.
- ^ Eyzen A (2002). "Amiotrofik lateral skleroz: sharh". BCMJ. 44 (7): 362-336. Arxivlandi asl nusxasi 2013 yil 21-iyun kuni. Olingan 21 avgust 2014.
- ^ Davenport RJ, Swingler RJ, kantsler AM, Warlow CP (fevral 1996). "Dvigatel neyron kasalligining soxta ijobiy tashxislaridan saqlanish: Shotlandiyaning motorli neyron kasalliklari registridan darslar". Nevrologiya, neyroxirurgiya va psixiatriya jurnali. 60 (2): 147–51. doi:10.1136 / jnnp.60.2.147. PMC 1073793. PMID 8708642.
- ^ Chieia MA, Oliveira AS, Silva HC, Gabbai AA (2010 yil dekabr). "Amiotrofik lateral skleroz: diagnostika mezonlari bo'yicha fikrlar". Arquivos de Neuro-Psiquiatria. 68 (6): 837–42. doi:10.1590 / S0004-282X2010000600002. PMID 21243238.
- ^ Al-Asmi A, Nandagopal R, Jacob PC, Gujjar A (2012 yil fevral). "Myasthenia Gravisning noto'g'ri diagnostikasi va undan keyingi klinik ta'siri: voqea hisoboti va adabiyotlarni ko'rib chiqish". Sulton Qobus universiteti tibbiy jurnali. 12 (1): 103–8. doi:10.12816/0003095. PMC 3286704. PMID 22375266.
- ^ a b v Abe K, Masashi A, Tsuji S, Itoyama Y, Sobue G, Togo M va boshq. (2017 yil iyul). "Amiotrofik lateral skleroz bilan aniq belgilangan bemorlarda edaravonning xavfsizligi va samaradorligi: randomizatsiyalangan, ikki tomonlama ko'r, platsebo nazorati ostida o'tkazilgan sinov". Lanset. Nevrologiya. 16 (7): 505–12. doi:10.1016 / S1474-4422 (17) 30115-1. PMID 28522181.
- ^ a b v Yeo CJ, Simmons Z (may, 2018). "Edaravoneni ALS kasalligi bilan muhokama qilish: AQSh nuqtai nazaridan axloqiy asos". Amiotrofik lateral skleroz va frontotemporal degeneratsiya. 19 (3–4): 167–72. doi:10.1080/21678421.2018.1425455. PMID 29334251. S2CID 4627647.
- ^ a b v d Orrell RW (2010). "Dvigatel neyron kasalligi: ALS va SMA davolashning tizimli sharhlari". Britaniya tibbiyot byulleteni. 93: 145–59. doi:10.1093 / bmb / ldp049. PMID 20015852.
- ^ a b Radunovich A, Annane D, Rafiq M, Brassington R, Mustfa N (6 oktyabr 2017). "Amiotrofik skleroz / motorli neyron kasalligi uchun mexanik shamollatish". Tizimli sharhlarning Cochrane ma'lumotlar bazasi. 10 (10): CD004427. doi:10.1002 / 14651858.CD004427.pub4. PMC 6485636. PMID 28982219.
- ^ a b v Ahmed R, Newcombe R, Piper A, Lyuis S, Yee B, Kiernan M, Grunstein R (aprel 2016). "Uyquning buzilishi va amiotrofik lateral sklerozda nafas olish funktsiyasi". Uyquga oid dorilarni ko'rib chiqish. 26: 33–42. doi:10.1016 / j.smrv.2015.05.007. PMID 26166297.
- ^ a b Eyzen A, Krieger C (2013 yil noyabr). "Amiotrofik lateral sklerozni boshqarishda axloqiy mulohazalar". Neyrobiologiyada taraqqiyot. 110: 45–53. doi:10.1016 / j.pneurobio.2013.05.05.001. PMID 23735671. S2CID 26282198.
- ^ a b Lyuis M, Rushanan S (2007). "Amiotrofik lateral sklerozni davolashda fizik terapiya va kasbiy terapiyaning ahamiyati". Neyro reabilitatsiya. 22 (6): 451–61. doi:10.3233 / NRE-2007-22608. PMID 18198431.
- ^ a b v d e "Amiotrofik lateral skleroz (ALS)". Amerikalik nutqni tinglash assotsiatsiyasi, Rokvill, MD. Arxivlandi asl nusxasi 2012 yil 2-avgustda. Olingan 30 noyabr 2016.
- ^ a b v Arbesman M, Sheard K (2014). "Amiotrofik lateral skleroz bilan kasallanganlar uchun kasbiy terapiya bilan bog'liq aralashuvlar samaradorligini tizimli ko'rib chiqish". Amerika kasbiy terapiya jurnali. 68 (1): 20–26. doi:10.5014 / ajot.2014.008649. PMID 24367951.
- ^ a b Katzberg HD, Benatar M (yanvar 2011). "Amiotrofik lateral skleroz / motorli neyron kasalligi uchun enteral naychani oziqlantirish". Tizimli sharhlarning Cochrane ma'lumotlar bazasi (1): CD004030. doi:10.1002 / 14651858.CD004030.pub3. PMC 7163276. PMID 21249659.
- ^ a b v Andersen PM, Abrahams S, Borasio GD, de Carvalho M, Chio A, Van Damm P va boshq. (2012 yil mart). "Amiotrofik lateral skleroz (MALS) klinik boshqaruvi bo'yicha EFNS ko'rsatmalari - EFNS ishchi guruhining qayta ko'rib chiqilgan hisoboti". Evropa nevrologiya jurnali. 19 (3): 360–75. doi:10.1111 / j.1468-1331.2011.03501.x. PMID 21914052. S2CID 5746940.
- ^ Carlesi C, Pasquali L, Piazza S, Lo Gerfo A, Caldarazzo Ienco E, Alessi R, Fornai F, Siciliano G (mart 2011). "Amiotrofik lateral sklerozda neyrodejeneratsiyaga klinik yondashuv strategiyasi". Italiya de Biologie arxivi. 149 (1): 151–67. doi:10.4449 / aib.v149i1.1267. PMID 21412722.
- ^ Hardiman O, van den Berg LH (iyul 2017). "Edaravone: ufqda ALS uchun yangi davolash?". Lanset. Nevrologiya. 16 (7): 490–91. doi:10.1016 / S1474-4422 (17) 30163-1. PMID 28522180. S2CID 7609862.
- ^ a b v d e f g Dorst J, Lyudolf A, Huebers A (2018). "Amiotrofik lateral sklerozni kasalliklarni o'zgartiruvchi va simptomatik davolash". Asab kasalliklarining terapevtik yutuqlari. 11: 1756285617734734. doi:10.1177/1756285617734734. PMC 5784546. PMID 29399045.
- ^ a b Takei K, Vatanabe K, Yuki S, Akimoto M, Sakata T, Palumbo J (oktyabr 2017). "Edaravone va uning amiotrofik lateral sklerozi uchun klinik rivojlanishi". Amiotrofik lateral skleroz. 18 (sup 1): 5-10. doi:10.1080/21678421.2017.1353101. PMID 28872907.
- ^ Paganoni, Sabrina; Xendrix, Suzanna; Dikson, Samuel P.; Nellton, Nyuman; Maklin, Erik A.; Berri, Jeyms D .; Elliott, Maykl A.; Mayser, Shomuil; Karam, Chafic; Caress, Jeyms B.; Owegi, Margaret Ayo (2020). "ALSda natriy fenilbutirat-taurursodiolni CENTAUR sinovida ishtirokchilarning uzoq muddatli omon qolishi". Mushak va asab. n / a (n / a). doi:10.1002 / mus.27091. ISSN 1097-4598. PMID 33063909.
- ^ Belluck, Pam. "Giyohvand moddalar ALS bemorlarining umrini bir necha oyga uzaytirishi mumkin". chicagotribune.com. Olingan 23 oktyabr 2020.
- ^ a b Paganoni S, Karam C, Joys N, Bedlack R, Carter G (2015). "Amiotrofik lateral skleroz spektri bo'yicha kompleks reabilitatsiya yordami". Neyro reabilitatsiya. 37 (1): 53–68. doi:10.3233 / NRE-151240. PMC 5223769. PMID 26409693.
- ^ Danel-Brunaud V, Touzet L, Chevalier L, Moreau C, Devos D, Vandoolaeghe S, Lefebvre L (2017 yil may). "Amiotrofik lateral skleroz bilan og'rigan bemorlarda axloqiy mulohazalar va palliativ yordam: sharh". Revue Neurologique. 173 (5): 300–07. doi:10.1016 / j.neurol.2017.03.032. PMID 28479121.
- ^ Checkoway H, Lundin JI, Kelada SN (2011). "22-bob: Neyrodejenerativ kasalliklar". Rotman N, Xaynaut P, Shulte P, Smit M, Boffetta P, Perera F (tahr.). Molekulyar epidemiologiya: asoslari va amaliyoti. Xalqaro saraton tadqiqotlari agentligi. 408-409 betlar. ISBN 978-9283221630.
- ^ Chiò A, Logroscino G, Traynor BJ, Collins J, Simeone JC, Goldstein LA, White LA (2013). "Amiotrofik lateral sklerozning global epidemiologiyasi: nashr etilgan adabiyotlarning tizimli sharhi". Neyroepidemiologiya. 41 (2): 118–30. doi:10.1159/000351153. PMC 4049265. PMID 23860588.
- ^ a b Artur KC, Calvo A, Price TR, Geiger JT, Chiò A, Traynor BJ (11 avgust 2016). "2015 yildan 2040 yilgacha amiotrofik lateral sklerozning prognozli o'sishi". Tabiat aloqalari. 7 (12408): 12408. Bibcode:2016 yil NatCo ... 712408A. doi:10.1038 / ncomms12408. PMC 4987527. PMID 27510634.
- ^ a b Luna J, Logroscino G, Couratier P, Marin B (may 2017). "ALS epidemiologiyasining dolzarb muammolari: populyatsiya va jismoniy faollik o'rtasida ALS paydo bo'lishining o'zgarishi xavf omilidir". Revue Neurologique. 173 (5): 244–53. doi:10.1016 / j.neurol.2017.03.035. PMID 28477849.
- ^ Zou ZY, Zhou ZR, Che CH, Liu CY, He RL, Huang HP (2017 yil iyul). "Amiotrofik lateral sklerozning genetik epidemiologiyasi: tizimli tahlil va meta-tahlil". Nevrologiya, neyroxirurgiya va psixiatriya jurnali. 88 (77): 540–49. doi:10.1136 / jnnp-2016-315018. PMID 28057713. S2CID 41974606.
- ^ Visser J, de Jong JM, de Visser M (2008 yil fevral). "Progressiv mushak atrofiyasi tarixi: sindrommi yoki kasallikmi?". Nevrologiya. 70 (9): 723–27. doi:10.1212 / 01.wnl.0000302187.20239.93. PMID 18299524. S2CID 22629725.
- ^ Wijesekera LC, Mathers S, Talman P, Galtrey C, Parkinson MH, Ganesalingam J, Willey E, Ampong MA, Ellis CM, Shaw CE, Al-Chalabi A, Leigh PN (mart 2009). "Tabiat tarixi va klinik qanotlari va yelka oyoqlari ALS variantlari". Nevrologiya. 72 (12): 1087–94. doi:10.1212 / 01.wnl.0000345041.83406.a2. PMC 2821838. PMID 19307543.
- ^ Li SE (2011 yil dekabr). "2011 yilda Guam demans sindromi qayta ko'rib chiqildi". Nevrologiyaning hozirgi fikri. 24 (6): 517–24. doi:10.1097 / WCO.0b013e32834cd50a. PMID 21986681.
- ^ Bruks BR, Sanjak M, Ringel S, Angliya J, Brinkmann J, Pestronk A va boshq. (1996 yil fevral). "Amiotrofik lateral sklerozning funktsional reyting shkalasi: amiotrofik lateral sklerozli bemorlarda kunlik hayot faoliyatini baholash". Nevrologiya arxivi. 53 (2): 141–7. doi:10.1001 / archneur.1996.00550020045014. PMID 8639063.
- ^ Cedarbaum JM, Stambler N, Malta E, Fuller C, Xilt D, Thurmond B, Nakanishi A (oktyabr 1999). "ALSFRS-R: nafas olish funktsiyasini baholashni o'z ichiga olgan qayta ishlangan ALS funktsional reyting shkalasi". Nevrologiya fanlari jurnali. 162 (1–2): 13–21. doi:10.1016 / s0022-510x (99) 00210-5. PMID 10540002. S2CID 7057926.
- ^ Wilbourn AJ (1998 yil oktyabr). "Amiotrofik lateral skleroz diagnostikasidagi klinik neyrofiziologiya: Lambert va El Eskaliy mezonlari". Nevrologiya fanlari jurnali. 160 (1-qo'shimcha): S25-29. doi:10.1016 / s0022-510x (98) 00194-4. PMID 9851644. S2CID 32884687.
- ^ Bruks BR (1994 yil iyul). "El Escorial Butunjahon Nevrologiya Federatsiyasi Amiotrofik Lateral Skleroz diagnostikasi mezonlari". Nevrologiya fanlari jurnali. 124 (Qo'shimcha): 96-107. doi:10.1016 / 0022-510x (94) 90191-0. PMID 7807156. S2CID 32678612.
- ^ Bruks BR, Miller RG, Swash M, Munsat TL (2000 yil dekabr). "El Escorial qayta ko'rib chiqildi: amyotrofik lateral skleroz diagnostikasi mezonlari qayta ko'rib chiqildi". Amiotrofik lateral skleroz va boshqa vosita neyron kasalliklari. 1 (5): 293–9. doi:10.1080/146608200300079536. PMID 11464847. S2CID 22725949.
- ^ a b Teive HA, Lima PM, Germiniani FM, Munhoz RP (may 2016). "Ism nima? Nervologik eponimlarga oid muammolar, faktlar va tortishuvlar". Arquivos de Neuro-Psiquiatria. 74 (5): 423–25. doi:10.1590 / 0004-282X20160040. PMID 27191240.
- ^ a b "ALS nima?". ALS assotsiatsiyasi. Arxivlandi asl nusxasidan 2018 yil 21 dekabrda. Olingan 23 dekabr 2018.
- ^ "ALS: lateral amiotrofik skleroz". Muskul distrofiyasi assotsiatsiyasi. 2015 yil 18-dekabr. Arxivlandi asl nusxasidan 2018 yil 6-avgustda. Olingan 23 dekabr 2018.
- ^ Goetz CG (2000 yil mart). "Amiotrofik lateral skleroz: Jan-Martin Sharkoning dastlabki hissalari". Mushak va asab. 23 (3): 336–43. doi:10.1002 / (SICI) 1097-4598 (200003) 23: 3 <336 :: AID-MUS4> 3.0.CO; 2-L. PMID 10679709.
- ^ Gordon PH (2006). "1-bob: ALS tarixi". Mitsumoto H, Przedborski S, Gordon PH (tahrir). Amiotrofik lateral skleroz. CRC Press. p. 9. ISBN 978-0824729240.
- ^ Mehta AR, Mehta PR, Anderson SP, MacKinnon BL, Compston A (yanvar 2020). "Kulrang modda etimologiyasi va neyron (e)". Miya. 143 (1): 374–379. doi:10.1093 / brain / awz367. PMC 6935745. PMID 31844876.
- ^ Gordon PH (oktyabr 2013). "Amiotrofik lateral skleroz: 2013 yil klinik xususiyatlari, patofiziologiyasi, boshqaruvi va terapevtik sinovlari uchun yangilanish". Qarish va kasalliklar. 4 (5): 295–310. doi:10.14336 / AD.2013.0400295. PMC 3794725. PMID 24124634.
- ^ a b Huynh V, Simon NG, Grosskreutz J, Turner MR, Vucic S, Kiernan MC (iyul 2016). "Amiotrofik lateral sklerozda yuqori motorli neyronni baholash". Klinik neyrofiziologiya. 127 (7): 2643–2660. doi:10.1016 / j.clinph.2016.04.025. PMID 27291884. S2CID 3757685.
- ^ "Motor neyron kasalliklari". www.uclh.nhs.uk. Olingan 26 oktyabr 2020.
- ^ "Jorj Bush, ehtimol ALS muz paqiridagi eng yaxshi chaqiruvni taqdim etadi". Mustaqil. Arxivlandi asl nusxasidan 2014 yil 21 avgustda. Olingan 20 avgust 2014.
- ^ "Ice Bucket Challenge ALS (MND) tadqiqotlarida gen kashfiyotini moliyalashtiradi". BBC yangiliklari. 2016 yil 27-iyul. Arxivlandi asl nusxasidan 2016 yil 28 iyuldagi. Olingan 27 iyul 2016.
- ^ Aleksandr, Ella. "Ice Bucket Challenge: Ledi Gaga, Jastin Biber, G-Dragon va Opra - hozirgacha eng qiziqarli reaktsiyalar".
- ^ "Ice Bucket Challenge ALS bilan bog'liq bo'lgan genni kashf qilishni mablag 'bilan ta'minladi. Arxivlandi asl nusxasidan 2016 yil 27 iyulda. Olingan 27 iyul 2016.
- ^ Kenna KP, van Doormaal PT, Dekker AM va boshq. (Sentyabr 2016). "NEK1 variantlari lateral amiotrofik sklerozga moyillikni beradi". Tabiat genetikasi. 48 (9): 1037–42. doi:10.1038 / ng. 3626. PMC 5560030. PMID 27455347.
- ^ a b v d e Moujalled D, White AR (mart 2016). "Amiotrofik lateral skleroz uchun kasalliklarni o'zgartirish usullarini ishlab chiqishdagi yutuqlar". CNS dorilar. 30 (3): 227–43. doi:10.1007 / s40263-016-0317-8. hdl:11343/216653. PMID 26895253. S2CID 13487502.
- ^ Picher-Martel V, Valdmanis PN, Gould PV, Julien JP, Dupré N (iyul 2016). "Hayvon modellaridan odam kasalligiga: ALSda shaxsiy tibbiyot uchun genetik yondashuv". Acta Neuropathologica Communications. 11 (4): 70. doi:10.1186 / s40478-016-0340-5. PMC 4940869. PMID 27400686.
- ^ a b Mitsumoto H, Bruks BR, Silani V (2014 yil noyabr). "Amiotrofik lateral sklerozdagi klinik tadqiqotlar: nega shuncha salbiy sinovlar va sinovlarni qanday yaxshilash mumkin?". Lanset. Nevrologiya. 13 (11): 1127–38. doi:10.1016 / S1474-4422 (14) 70129-2. PMID 25316019. S2CID 28510979.
- ^ Fang J, Chjou M, Yang M, Chju S, Xe L (may, 2013). "Amiotrofik lateral skleroz yoki motorli neyron kasalligini davolash uchun takroriy transkranial magnit stimulyatsiya". Tizimli sharhlarning Cochrane ma'lumotlar bazasi (5): CD008554. doi:10.1002 / 14651858.CD008554.pub3. PMC 7173713. PMID 23728676.
- ^ Chen KS, Sakowski SA, Feldman EL (mart 2016). "Amiotrofik lateral skleroz uchun intraspinal ildiz hujayralarini transplantatsiyasi". Nevrologiya yilnomalari. 79 (3): 342–53. doi:10.1002 / ana.24584. PMC 4789073. PMID 26696091.
- ^ Abdul Vohid, S. Fadila; Qonun, Zhe Kang; Ismoil, Nor Azima; Lay, Nai Ming (2019 yil 19-dekabr). "Amiotrofik lateral skleroz / motorli neyron kasalligi uchun hujayralar asosida davolash usullari". Tizimli sharhlarning Cochrane ma'lumotlar bazasi. 12: CD011742. doi:10.1002 / 14651858.CD011742.pub3. ISSN 1469-493X. PMC 6920743. PMID 31853962.
- ^ "Amitrofik lateral sklerozni davolash uchun" Masitinib mesilat "ni etim nomi bilan belgilash bo'yicha jamoatchilik fikri" (PDF). EMA. Evropa dori-darmon agentligi, etimlarga tegishli tibbiy mahsulotlar qo'mitasi 22 sentyabr 2016 yil. Arxivlandi (PDF) asl nusxasidan 2016 yil 6-noyabrda. Olingan 6 noyabr 2016.
- ^ Bartus RT, Bétourné A, Basile A, Peterson BL, Glass J, Boulis NM (yanvar 2016). "b2-adrenoseptor agonistlari amiotrofik lateral skleroz (ALS) uchun yangi, xavfsiz va potentsial samarali terapiya sifatida". Kasallikning neyrobiologiyasi. 85: 11–24. doi:10.1016 / j.nbd.2015.10.006. PMID 26459114. S2CID 536279.
- ^ Bräuer S, Zimyanin V, Hermann A (aprel 2018). "Amiotrofik lateral sklerozda kasallikka tegishli oqsillarning prionga o'xshash xususiyatlari". Asab uzatish jurnali. 125 (4): 591–613. doi:10.1007 / s00702-018-1851-y. PMID 29417336. S2CID 3895544.
- ^ Lau DH, Hartopp N, Welsh NJ, Myuller S, Glennon EB, Mórotz GM, Annibali A, Gomez-Suaga P, Stoica R, Paillusson S, Miller CC (2018 yil fevral). "Front-temporal demans va unga bog'liq amiotrofik lateral sklerozda ER-mitoxondriya signalizatsiyasining buzilishi". Hujayra o'limi va kasallik. 9 (3): 327. doi:10.1038 / s41419-017-0022-7. PMC 5832427. PMID 29491392.
- ^ Neumann M (oktyabr 2013). "Frontotemporal lobar degeneratsiyasi va amiotrofik lateral skleroz: molekulyar o'xshashlik va farqlar". Revue Neurologique. 169 (10): 793–98. doi:10.1016 / j.neurol.2013.07.019. PMID 24011641.
Tashqi havolalar
| Tasnifi | |
|---|---|
| Tashqi manbalar |
| Vakolat nazorati |
|---|