Huntington kasalligi - Huntingtons disease - Wikipedia
Ushbu maqola bo'lishi kerak yangilangan. (2020 yil mart) |
| Xantington kasalligi | |
|---|---|
| Boshqa ismlar | Xantington xoreyasi |
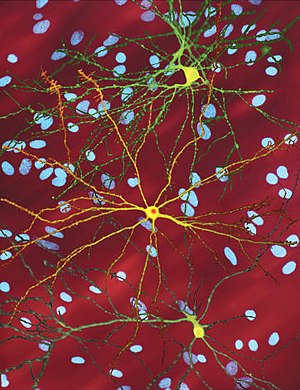 | |
| A-ning tahrirlangan mikroskopik tasviri o'rta mayda neyron (sariq) bilan inklyuziya tanasi (to'q sariq), bu kasallik jarayonining bir qismi sifatida yuzaga keladi (rasm kengligi 360µm ) | |
| Mutaxassisligi | Nevrologiya |
| Alomatlar | Muvofiqlashtirish va yurish, kayfiyat va aqliy qobiliyatlarni o'z ichiga olgan vosita mahoratiga oid muammolar[1][2] |
| Asoratlar | Zotiljam, yurak kasalligi, qulab tushganda jismoniy shikastlanish, o'z joniga qasd qilish[3] |
| Odatiy boshlanish | 30-50 yoshda[4] |
| Muddati | Uzoq muddat[4] |
| Sabablari | Genetik (irsiy yoki yangi mutatsiya)[4] |
| Diagnostika usuli | Genetik sinov[5] |
| Differentsial diagnostika | Sydenhamning xoresi, benign irsiy xoreya, lupus, paraneoplastik sindrom, Uilson kasalligi[6] |
| Davolash | Qo'llab-quvvatlash[2] |
| Dori-darmon | Tetrabenazin[3] |
| Prognoz | Tashxis qo'yilganidan 15-20 yil o'tgach[4] |
| Chastotani | 100000 yilda 4-15 (Evropa kelib chiqishi)[1] |
Xantington kasalligi (HD), shuningdek, nomi bilan tanilgan Xantington xoreyasi, a neyrodejenerativ kasallik bu asosan meros qilib olingan.[7] Dastlabki alomatlar ko'pincha kayfiyat yoki aqliy qobiliyatlar bilan bog'liq nozik muammolardir.[1] Umumiy muvofiqlashtirishning etishmasligi va beqaror yurish tez-tez kuzatib boring.[2] Kasallik avjga chiqqanda, taniqli muvofiqlashtirilmagan, beixtiyor harakatlar xorea yanada ravshanroq bo'lish.[1] Jismoniy qobiliyatlar tobora yomonlashib boradi muvofiqlashtirilgan harakat qiyinlashadi va odam gapira olmaydi.[1][2] Aqliy qobiliyatlar odatda pasayish dementia.[3] Odamlar orasida o'ziga xos alomatlar biroz farq qiladi.[1] Alomatlar odatda 30 yoshdan 50 yoshgacha boshlanadi, ammo har qanday yoshda boshlanishi mumkin.[4][3] Kasallik har bir keyingi avlodda hayotdan oldin rivojlanishi mumkin.[1] Taxminan sakkiz foiz holatlar 20 yoshdan oldin boshlanadi va ular sifatida tanilgan voyaga etmaganlar uchun HD, odatda bilan ko'rsatilgan sekin harakatlanish belgilari ning Parkinson kasalligi xoreya emas.[3]
HD odatda ta'sirlangan ota-onadan meros qilib olingan, kim ko'taradi mutatsiya ichida ov qiluvchi gen (HTT).[4] Ammo, 10% gacha bo'lgan holatlar yangi mutatsiyaga bog'liq.[1] Huntingtin geni uchun genetik ma'lumot beradi ovtin oqsili (htt).[1] Kengayishi CAG takrorlaydi ning sitozin -adenin -guanin (a nomi bilan tanilgan trinukleotidning takroriy kengayishi ) Huntin oqsili uchun kodlash genida g'ayritabiiy mutant oqsil (mhtt) paydo bo'lib, u asta-sekin zarar etkazadi miya hujayralari bir qator mumkin bo'lgan mexanizmlar orqali.[7][8] Tashxis qo'yilgan genetik test, bu alomatlar mavjud bo'lishidan yoki yo'qligidan qat'i nazar, har qanday vaqtda amalga oshirilishi mumkin.[5] Bu haqiqat bir nechta axloqiy munozaralarni keltirib chiqaradi: test sinovlarini tanlash uchun shaxsning etuk deb hisoblangan yoshi; ota-onalar farzandlarini tekshiruvdan o'tkazish huquqiga egami; va maxfiylikni boshqarish va test natijalarini oshkor qilish.[2]
HD-ni davolash mumkin emas va keyingi bosqichlarda to'la vaqtli parvarish talab etiladi.[2] Davolash usullari ba'zi alomatlarni engillashtirishi va ba'zilarida yaxshilanishi mumkin hayot sifati.[3] Harakat muammolarini davolash uchun eng yaxshi dalillar tetrabenazin.[3] Evropadan kelib chiqqan 100000 odamda HD taxminan 4 dan 15 gacha ta'sir qiladi.[1][3] Bu yaponlar orasida kamdan-kam uchraydi, Afrikada paydo bo'lish darajasi esa noma'lum.[3] Kasallik erkaklar va ayollarga teng ta'sir qiladi.[3] Kabi asoratlar zotiljam, yurak kasalligi va tushishdan jismoniy shikastlanish umr ko'rish davomiyligini pasaytiradi.[3] O'z joniga qasd qilish taxminan 9% hollarda o'limga sabab bo'ladi.[3] O'lim odatda kasallik birinchi marta aniqlangandan 15-20 yil o'tgach sodir bo'ladi.[4]
Kasallikning birinchi ehtimol tavsifi 1841 yilda amerikalik shifokor Charlz Oskar Uoters tomonidan qilingan.[9] Vaziyat 1872 yilda amerikalik shifokor tomonidan batafsilroq tavsiflangan Jorj Xantington.[9] Genetika asoslari 1993 yilda boshchiligidagi xalqaro birgalikdagi sa'y-harakatlar bilan aniqlandi Herediter Disease Foundation.[10][11] Tadqiqot va qo'llab-quvvatlovchi tashkilotlar jamoatchilikni xabardorligini oshirish, shaxslar va ularning oilalarini qo'llab-quvvatlash va tadqiqotlarni rivojlantirish uchun 1960-yillarning oxirida shakllana boshladi.[12][11] Tadqiqot yo'nalishlari kasallikning aniq mexanizmini aniqlash, takomillashtirishni o'z ichiga oladi hayvon modellari tadqiqotlarga yordam berish, kasallik alomatlarini davolash yoki sekinlashishini davolash uchun dori-darmonlarni sinash va shunga o'xshash protseduralarni o'rganish ildiz hujayralari terapiyasi shikastlangan yoki yo'qolgan neyronlarni almashtirish maqsadi bilan.[10]
Belgilari va alomatlari
| Jahldorlik | 38–73% |
| Apatiya | 34–76% |
| Tashvish | 34–61% |
| Tushkun kayfiyat | 33–69% |
| Obsesif va majburiy | 10–52% |
| Psixotik | 3–11% |
Xantington kasalligining alomatlari odatda 30 yoshdan 50 yoshgacha seziladi, ammo ular har qanday yoshda boshlanishi mumkin.[4] Ularning rivojlanishi ko'pincha erta bosqichlarda, o'rta bosqichlarda va oldingi prodromal faza bilan kech bosqichlarda tavsiflanadi.[2] Dastlabki bosqichlarda shaxsiyatning nozik o'zgarishlari, muammolari mavjud bilish va jismoniy ko'nikmalar, asabiylashish va kayfiyatning o'zgarishi, bularning barchasi e'tiborga olinmasligi mumkin,[14][15] va bu odatda vosita alomatlaridan oldin.[16] HD bilan deyarli har bir kishi oxir-oqibat o'xshash jismoniy alomatlarni namoyon qiladi, ammo kognitiv va xulq-atvor belgilarining paydo bo'lishi, rivojlanishi va darajasi individual ravishda sezilarli darajada farq qiladi.[17][18]
Eng xarakterli dastlabki jismoniy alomatlar - bu siltang, tasodifiy va boshqarib bo'lmaydigan harakatlar xorea.[19] Ko'p odamlar o'zlarining beixtiyor harakatlaridan xabardor emaslar yoki ularga to'sqinlik qilmoqdalar.[1] Xoreya dastlab umumiy bezovtalik, istalmagan tarzda boshlangan yoki tugallanmagan kichik harakatlar, muvofiqlashtirishning etishmasligi yoki sekinlashuv sifatida namoyon bo'lishi mumkin. sakkadik ko'z harakatlari.[19] Ushbu kichik motor anormalliklari, odatda, kamida uch yilgacha vosita buzilishining aniq belgilaridan oldin bo'ladi.[17] Qattiqlik, burish harakatlari yoki kabi belgilarning aniq ko'rinishi g'ayritabiiy posturing buzilish rivojlanib borishi bilan paydo bo'ladi.[19] Bular harakatga javob beradigan miyadagi tizim ta'sirlanganligining alomatlaridir.[20] Psixomotor funktsiyalar tobora zaiflashib boradi, shuning uchun mushaklarni nazorat qilishni talab qiladigan har qanday harakat ta'sir qiladi. Umumiy oqibatlar - jismoniy beqarorlik, yuzning g'ayritabiiy ifodasi va chaynashdagi qiyinchiliklar, yutish va Gapirmoqda.[19] Uyquning buzilishi va Ozish ham bog'liq simptomlardir.[21] Ovqatlanishda qiyinchiliklar odatda vazn yo'qotishiga olib keladi va to'yib ovqatlanishga olib kelishi mumkin.[22][23] Voyaga etmaganlarning HD darajasi, kognitiv pasayish bilan tezroq tezroq rivojlanadi va xoreya qisqacha namoyish etiladi; The Vestfal varianti ning harakatning sustligi, qattiqlik va silkinishlar voyaga etmaganlarning HD-da odatdagidek ko'proq uchraydi soqchilik.[19][21]
Kognitiv qobiliyat asta-sekin buziladi.[20] Ayniqsa, ta'sirlanganlar ijro funktsiyalari rejalashtirish, bilim moslashuvchanligi, mavhum fikrlash, qoidalarni sotib olish, tegishli harakatlarni boshlash va noo'rin harakatlarning oldini olish.[20] Kasallik o'sib borishi bilan xotira defitsitlar paydo bo'lishga moyildir. Hisobotdagi buzilishlar oralig'ida qisqa muddatli xotira defitsit uzoq muddatli xotira qiyinchiliklar, jumladan, defitsit epizodik (o'z hayotining xotirasi), protsessual (qanday faoliyatni amalga oshirish kerakligi haqida tananing xotirasi) va ishlaydigan xotira.[20] Kognitiv muammolar vaqt o'tishi bilan yomonlashishga moyil bo'lib, oxir-oqibat bunga olib keladi dementia.[20]
Xabar berildi asab-psixiatrik belgilar mavjud tashvish, depressiya, a his-tuyg'ularning pasayishi, egosentrizm, tajovuz va majburiy xatti-harakatlar, ikkinchisi sabab bo'lishi yoki yomonlashishi mumkin giyohvandlik, shu jumladan alkogolizm, qimor va giperseksualizm.[13] Boshqalarning salbiy ifodalarini tan olishdagi qiyinchiliklar ham kuzatilgan.[20] The tarqalishi Ushbu alomatlar tadqiqotlar orasida juda o'zgaruvchan bo'lib, umr bo'yi tarqalishining taxminiy darajasi psixiatrik kasalliklar 33% dan 76% gacha.[13] Ko'pgina azob chekuvchilar va ularning oilalari uchun ushbu alomatlar kasallikning eng xavotirli tomonlaridan biri bo'lib, ko'pincha kundalik ishlashga ta'sir qiladi va sabab bo'ladi. institutsionalizatsiya.[13] O'z joniga qasd qilish fikri va o'z joniga qasd qilish urinishlari umumiy aholiga qaraganda tez-tez uchraydi.[19] Ko'pincha shaxslar xoreya, kognitiv va hissiy nuqsonlar to'g'risida xabardorlikni pasaytirdilar.[24]
Mutant ovlanish butun tanada namoyon bo'ladi va periferik to'qimalarda anormallik bilan bog'liq bo'lib, ular bevosita miyadan tashqarida bunday ifoda tufayli yuzaga keladi. Ushbu anormalliklarga quyidagilar kiradi mushak atrofiyasi, yurak etishmovchiligi, buzilgan glyukoza bardoshlik, Ozish, osteoporoz va moyak atrofiyasi.[25]
Genetika
Har kimning ikki nusxasi bor ov qiluvchi gen (HTT), qaysi uchun kodlar ovtin oqsili (htt). HTT ham HD gen, va IT15 geni, (qiziqarli stenogramma 15). Ushbu genning bir qismi takroriy bo'lim bo'lib, a deb nomlanadi trinukleotidning takroriy kengayishi - a qisqa takrorlash jismoniy shaxslar o'rtasida uzunligi o'zgarib turadi va avlodlar o'rtasidagi uzunlikni o'zgartirishi mumkin. Agar takrorlanish sog'lom genda mavjud bo'lsa, dinamik mutatsiya takrorlanish sonini ko'paytirishi va nuqsonli genga olib kelishi mumkin. Ushbu takrorlanadigan qismning uzunligi ma'lum bir chegaraga etganida, u mutant huntin oqsili (mhtt) deb nomlangan o'zgargan oqsil shaklini hosil qiladi. Ushbu oqsillarning turli funktsiyalari patologik o'zgarishlarning sababi bo'lib, ular kasallik alomatlarini keltirib chiqaradi. Xantington kasalligining mutatsiyasi genetik jihatdan dominant bo'lib, deyarli to'liq kiruvchi: biron bir kishining mutatsiyasi HTT allellar kasallikka sabab bo'ladi. U jinsga ko'ra meros qilib olinmaydi, lekin genning takroriy bo'limi uzunligi va shu sababli uning zo'ravonligiga ta'sirlangan ota-onaning jinsi ta'sir qilishi mumkin.[19]
Genetik mutatsiya
HD - bu bir nechta narsalardan biri trinukleotidni takroriy buzilishi ular genning takroriy kesimining normal diapazondan oshib ketishidan kelib chiqadi.[19] The HTT gen joylashgan kalta qo'l ning xromosoma 4[19] 4p16.3 da. HTT uchta ketma-ketlikni o'z ichiga oladi DNK asoslari - sitozin-adenin-guanin (CAG) - trinukleotid takrorlanishi deb nomlanuvchi bir necha marta takrorlangan (ya'ni ... CAGCAGCAG ...).[19] CAG - bu uchta harf genetik kod (kodon ) uchun aminokislota glutamin, shuning uchun ularning ketma-ketligi a deb nomlanuvchi glutamin zanjirini ishlab chiqarishga olib keladi poliglutamin trakti (yoki polyQ trakti) va genning takrorlangan qismi PolyQ mintaqasi.[26]
| Hisobni takrorlang | Tasnifi | Kasallik holati | Avlod uchun xavf |
|---|---|---|---|
| <27 | Oddiy | Ta'sir qilinmaydi | Yo'q |
| 27–35 | O'rta | Ta'sir qilinmaydi | Ko'tarilgan, ammo <50% |
| 36–39 | Nafas qisqartirildi | Ta'sir qilishi mumkin yoki bo'lmasligi mumkin | 50% |
| 40+ | To'liq Penetrance | Ta'sir qilinadi | 50% |
Odatda, odamlarda polyQ mintaqasida 36 dan kam takrorlangan glutamin mavjud bo'lib, natijada ular ishlab chiqariladi sitoplazmatik oqsil ovlash.[19] Shu bilan birga, 36 yoki undan ortiq glutaminlar ketma-ketligi natijasida turli xil xususiyatlarga ega bo'lgan oqsil ishlab chiqariladi.[19] Mutant huntingin (mhtt) deb nomlangan ushbu o'zgargan shakl ba'zi turdagi parchalanish tezligini oshiradi neyronlar. Miyaning mintaqalari turli xil miqdordagi va ushbu turdagi neyronlarga bog'liq bo'lib, ularga ta'sir qiladi.[19] Odatda, CAG takrorlanishining soni bu jarayonga qanchalik ta'sir ko'rsatishi bilan bog'liq bo'lib, simptomlar paydo bo'lish yoshining o'zgarishi taxminan 60% ni tashkil qiladi. Qolgan o'zgarish HD muhitini o'zgartiradigan atrof-muhit va boshqa genlarga tegishli.[19] 36 dan 39 gacha takrorlash kasallikning penetran shaklini pasayishiga olib keladi, simptomlar ancha kech boshlanadi va sekinlashadi. Ba'zi hollarda boshlanish shu qadar kech bo'lishi mumkinki, alomatlar hech qachon sezilmaydi.[19] Juda katta takroriy hisoblashlar bilan (60 dan ortiq) HD paydo bo'lishi 20 yoshdan kichik bo'lib sodir bo'lishi mumkin voyaga etmaganlar uchun HD. Voyaga etmaganlar HD odatda Vestfal varianti bu harakatning sustligi, qat'iylik va titroq bilan tavsiflanadi. Bu HD tashuvchilarning taxminan 7 foizini tashkil qiladi.[27][28]
Meros olish
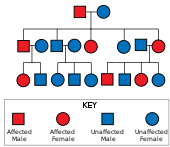
Xantington kasalligi bor autosomal dominant meros, ya'ni ta'sirlangan odam odatda genning bitta nusxasini kengaytirilgan trinukleotid takrorlanishi (mutant) bilan olishini anglatadi allel ) ta'sirlangan ota-onadan.[19] Mutatsiyaning penetratsiyasi juda yuqori bo'lganligi sababli, genning mutatsiyalangan nusxasiga ega bo'lganlar kasallikka chalinadi. Ushbu turdagi meros modelida zarar ko'rgan shaxsning har bir nasli mutant allelni meros qilib olish xavfi 50% ni tashkil qiladi va shuning uchun buzilish ta'sir qiladi (rasmga qarang). Bunday ehtimollik jinsiy aloqaga bog'liq emas.[29]
Trinukleotid CAG takrorlanadi davomida 28 dan ortiq beqaror takrorlash va bu beqarorlik mavjud takrorlanishlar sonining ko'payishi bilan ortadi.[19] Bu odatda avlodlar o'tishi bilan yangi kengayishlarga olib keladi (dinamik mutatsiyalar ) trinukleotid takrorlanishining aniq nusxasini ko'paytirish o'rniga.[19] Bu ketma-ket nasllarda takrorlanishlar sonining o'zgarishiga olib keladi, masalan, "oraliq" sonli takrorlanishga ega bo'lgan (28-35) yoki "penetrani kamaygan" (36-40) ota-ona gen nusxasini o'tishi mumkin. to'liq penetran HD ishlab chiqaradigan takroriy sonlarning ko'payishi bilan.[19] Takrorlash sonining bunday ko'payishi (va shuning uchun oldinroq) boshlanish yoshi va kasallikning og'irligi) ketma-ket nasllarda genetik deb nomlanadi kutish.[1] Beqarorlik ko'proq spermatogenez dan oogenez;[19] ona tomonidan meros bo'lib o'tgan allellar odatda takrorlanadigan uzunlikka ega, otadan meros bo'lib o'tganlarda esa uzunlikni oshirish ehtimoli katta.[19][30] Xantington kasalligi a sabab bo'lishi kamdan-kam uchraydi yangi mutatsiya, bu erda hech bir ota-onada 36 dan ortiq CAG takrorlanishi mavjud emas.[31]
Ikkala ota-ona ham kengaytirilgan HD geniga ega bo'lgan kamdan-kam holatlarda xavf 75% gacha ko'tariladi va agar ota-onalardan biri ikkita kengaytirilgan nusxaga ega bo'lsa, bu xavf 100% ni tashkil qiladi (barcha bolalar ta'sir qiladi). Jismoniy shaxslar ikkala gen ham ta'sir qildi kamdan-kam uchraydi. Bir muncha vaqt HD mutatsiyaga uchragan ikkinchi genga ega bo'lish simptomlar va o'sishga ta'sir qilmaydigan yagona kasallik deb o'ylardi,[32] ammo keyinchalik ta'sir qilishi mumkinligi aniqlandi fenotip va rivojlanish darajasi.[19][33]
Mexanizmlar
Huntingtin oqsili 100 dan ortiq boshqa oqsillar bilan o'zaro ta'sir qiladi va ko'p funktsiyalarga ega.[34] Mutatsiyaga uchragan oqsilning (mhtt) xatti-harakatlari to'liq tushunilmagan, ammo u hujayralarning ayrim turlari, xususan miyada zaharli hisoblanadi. Erta zarar eng aniq ko'rinishda striatum, ammo kasallik rivojlanib borishi bilan miyaning boshqa sohalari ham sezilarli darajada ta'sirlanadi. Dastlabki alomatlar striatum funktsiyalari va uning kortikal birikmalariga, ya'ni harakatni, kayfiyatni va yuqori kognitiv funktsiyalarni boshqarish bilan bog'liq.[19] DNK metilatsiyasi HD formatida ham o'zgartirilgan ko'rinadi.[35]
Huntingtin funktsiyasi
Huntingtin (HTT) bu ifoda etilgan miyada topilgan eng yuqori konsentratsiyalar bilan barcha hujayralarda moyaklar, va o'rtacha miqdor jigar, yurak va o'pka. Biroq, uning funktsiyasi aniq emas.[19] U transkripsiyada ishtirok etadigan oqsillar bilan o'zaro ta'sir qiladi, hujayra signalizatsiyasi va hujayra ichidagi tashish.[19][36] Hayvonlarda genetik jihatdan o'zgartirilgan HDni namoyish qilish uchun HTT ning bir nechta funktsiyalari aniqlandi.[37] Ushbu hayvonlarda HTT embrional rivojlanish uchun muhimdir, chunki uning yo'qligi embrion o'limi bilan bog'liq. Kaspaz, apoptozni katalizatsiyalashda rol o'ynaydigan ferment, mutatsiyalangan gen tomonidan ubiqitin-proteaz tizimiga zarar etkazish orqali faollashadi deb o'ylashadi. Shuningdek, u anti-apoptotik agentning oldini olish dasturlashtirilgan hujayralar o'limi va ishlab chiqarishni nazorat qiladi miyadan kelib chiqadigan neyrotrofik omil, neyronlarni himoya qiluvchi va ularning yaratilishini tartibga soluvchi oqsil neyrogenez. HTT ham osonlashtiradi vesikulyar transport va sinaptik uzatish va neyronlarning gen transkripsiyasini boshqaradi.[37] Agar ifoda HTT ko'paytirildi va ko'proq HTT ishlab chiqarildi, miya hujayrasi omon qolish yaxshilanadi va mhttning ta'siri kamayadi, HTT ekspressioni kamayganda esa natijada paydo bo'lgan xususiyatlar mhtt ishtirokida ko'rinadi.[37] Shunga ko'ra kasallik sabab bo'lmaydi deb o'ylashadi yetarli darajada ishlab chiqarilmaganligi HTT, lekin a tomonidan zaharli funktsiya funktsiyasi tanadagi mhtt[19]
Uyali o'zgarishlar
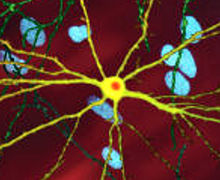
Mhttning toksik ta'siri HD patologiyasini namoyon qilishi va keltirib chiqarishi mumkin bo'lgan ko'plab uyali o'zgarishlar mavjud.[38][39] Mutant (ya'ni kengaytirilgan poliglutamin) shaklida oqsil parchalanishga moyil bo'lib, u poliglutamin kengayishini o'z ichiga olgan qisqaroq bo'laklar hosil qiladi.[38] Ushbu oqsil parchalari moyillikka ega noto'g'ri va ko'p miqdordagi oqsillardan hosil bo'lgan mahalliy bo'lmagan poliglutamin b-iplari vodorod aloqalari orqali bog'langan fibrillyar agregatlarni hosil qiladi.[8] Ushbu agregatlar bir xil asosiy o'zaro faoliyat qiymatga ega amiloid boshqa oqsillarni cho'ktirish kasalliklarida ko'rinadigan arxitektura. Vaqt o'tishi bilan agregatlar hosil bo'lish uchun to'planadi inklyuziya organlari hujayralar ichida, natijada neyronlarning ishlashiga xalaqit beradi.[38][8] Neyron qo'shilishlari bilvosita aralashuvga olib keladi. Inklyuziv organlar ikkalasida ham topilgan hujayra yadrosi va sitoplazma.[38] Miyaning hujayralariga qo'shilish tanalari dastlabki patologik o'zgarishlardan biri bo'lib, ba'zi tajribalar ular bo'lishi mumkinligini aniqladi zaharli hujayra uchun, ammo boshqa tajribalar shuni ko'rsatdiki, ular tanani himoya qilish mexanizmining bir qismi sifatida shakllanishi va hujayralarni himoya qilishga yordam berishi mumkin.[38]
Mhtt hujayraning o'limiga olib kelishi mumkin bo'lgan bir necha yo'llar aniqlandi. Bunga quyidagilar kiradi: ta'sirlar chaperone oqsillari, bu oqsillarni katlamaga va noto'g'ri katlanmışlarni olib tashlashga yordam beradi; bilan o'zaro aloqalar kaspalar da rol o'ynaydigan hujayralarni olib tashlash jarayoni; The glutaminning asab hujayralariga toksik ta'siri; hujayralar ichida energiya ishlab chiqarishning buzilishi; va genlarning ekspressioniga ta'siri.[8][40]
Mutant ov qilish oqsilida muhim rol o'ynashi aniqlandi mitoxondrial disfunktsiya.[41] Mitokondriyaning buzilishi elektron transport yuqori darajalarga olib kelishi mumkin oksidlovchi stress va ozod qilish reaktiv kislorod turlari.[42]
Glutamin ma'lum eksitotoksik ko'p miqdorda bo'lganda va eksitotoksinlar ko'plab uyali tuzilmalarga zarar etkazadi. Glutamin HD darajasida haddan tashqari ko'p miqdorda topilmaydi, ammo o'zgargan ovtin oqsilining neyronlardagi ko'p miqdordagi oqsillar bilan o'zaro ta'siri glutamin ta'sirchanligini kuchayishiga olib keladi. Zaiflikning oshishi normal glutamin darajasidan eksitotoksik ta'sirga olib keladi deb taxmin qilingan.[8]
Makroskopik o'zgarishlar

HD butun miyaga ta'sir qiladi, ammo ba'zi joylar boshqalarga qaraganda zaifroq. Eng ko'zga ko'ringan dastlabki effektlar bazal ganglionlar deb nomlangan striatum dan tashkil topgan kaudat yadrosi va putamen.[19] Ta'sir qilingan boshqa joylarga quyidagilar kiradi substantia nigra, kortikal qatlamlar 3, 5 va 6 ning neokorteks, gipokampus, Purkinje hujayralari ichida serebellum, ning lateral tuberal yadrolari gipotalamus va qismlari talamus.[19] Ushbu joylar ularning tuzilishiga va tarkibidagi neyronlarning turlariga qarab ta'sirlanib, hujayralarni yo'qotganda hajmi kamayadi.[19] Striatal o'rta tikanli neyronlar eng zaif, ayniqsa ular bilan himoyalangan proektsiyalar tomonga tashqi globus pallidus, bilan internironlar va proektsiyalangan tikanli hujayralar ichki globus pallidus kamroq ta'sirlanish.[19][43] HD ham sabab bo'ladi g'ayritabiiy o'sish yilda astrotsitlar va miyaning immunitet hujayralarining faollashishi, mikrogliya.[44]
Bazal ganglionlar - miyaning HD-ning boshida eng ko'p ta'sirlanadigan qismi harakat va xatti-harakatlarni boshqarishda muhim rol o'ynaydi. Ularning funktsiyalari to'liq tushunilmagan, ammo hozirgi nazariyalar ularni kognitivning bir qismi deb taxmin qilmoqda ijro etuvchi tizim[20] va dvigatel davri.[45] Bazal ganglionlar odatiy harakatlarni keltirib chiqaradigan ko'plab davrlarni inhibe qiladi. Muayyan harakatni boshlash uchun miya yarim korteksi bazal ganglionlarga signal yuboradi, bu esa inhibatsiyani chiqarishga olib keladi. Bazal ganglionlarga zarar etkazish inhibisyonlarning chiqarilishi yoki tiklanishining tartibsiz va nazoratsiz bo'lishiga olib kelishi mumkin, bu esa harakatlarning noqulay boshlanishiga yoki harakatlarning bexosdan boshlanishiga yoki harakatni uning tugatilishidan oldin yoki undan oldin to'xtatilishiga olib keladi. Ushbu sohada to'plangan zarar HD deb nomlanuvchi xarakterli notekis harakatlarni keltirib chiqaradi xorea, a diskinezi.[45] Bazal ganglionlarning harakatlarni to'xtata olmasligi sababli, ta'sirlangan shaxslar nutqni ishlab chiqarish va oziq-ovqat va suyuqliklarni (disfagiya) ishlab chiqarish qobiliyatini pasayishi muqarrar.[46]
Transkripsiya bilan tartibga solish
CREB bilan bog'langan oqsil (CBP), transkripsiya yadrosi, hujayraning ishlashi uchun juda muhimdir, chunki ko'plab promotorlarda koaktivator bo'lib, u omon qolish yo'llari uchun genlarning transkripsiyasini faollashtiradi.[40] Bundan tashqari, CBP hosil qiluvchi aminokislotalar tarkibiga 18 ta glutaminli tasma kiradi. Shunday qilib, CBPdagi glutaminlar to'g'ridan-to'g'ri HTT zanjiridagi ko'paygan glutamin bilan o'zaro ta'sir qiladi va CBP yadro yonidagi odatiy joyidan uzoqlashadi.[47] Xususan, CBP tarkibida atsetiltransferaza domeni mavjud bo'lib, unga HTT o'zining poliglutamin o'z ichiga olgan domeni orqali bog'lanadi.[48] Xantington kasalligiga chalinganlarning otopsi qilingan miyalarida KBP miqdori nihoyatda kamayganligi aniqlandi.[47] Bundan tashqari, CBP haddan tashqari ta'sirlanganda, poliglutamin tomonidan kelib chiqadigan o'lim kamayadi, bu esa CBP ning Xantington kasalligi va umuman neyronlarda muhim rol o'ynayotganligini namoyish etadi.[40]
Tashxis
Tibbiy tashxis HD boshlanishi kasallikka xos bo'lgan jismoniy alomatlar paydo bo'lgandan keyin amalga oshirilishi mumkin.[19] Genetik sinov agar oilada HD tarixi bo'lmasa, jismoniy tashxisni tasdiqlash uchun ishlatilishi mumkin. Alomatlar paydo bo'lishidan oldin ham, genetik tekshiruv individual yoki yo'qligini tasdiqlashi mumkin embrion trinukleotid takrorlanishining (CAG) kengaytirilgan nusxasini HTT kasallikni keltirib chiqaradigan gen. Genetik maslahat test jarayoni davomida va tasdiqlangan tashxisning oqibatlari to'g'risida maslahat va ko'rsatmalar berish uchun mavjud. Ushbu ta'sirlar shaxsning psixologiyasiga, martaba, oilani rejalashtirish qarorlari, qarindoshlari va munosabatlariga ta'sirini o'z ichiga oladi. Semptomatik tekshiruvlar mavjudligiga qaramay, HDni meros qilib olish xavfi bo'lganlarning atigi 5% i buni tanlaydi.[19]
Klinik

A fizik tekshiruv, ba'zan a bilan birlashtiriladi psixologik tekshirish, kasallikning boshlanishi boshlanganligini aniqlashi mumkin.[19] Tananing biron bir qismining ortiqcha bexosdan harakatlari ko'pincha tibbiy maslahat olish uchun sababdir. Agar ular keskin va tasodifiy vaqt va taqsimotga ega bo'lsa, ular HD tashxisini taklif qilishadi. Kognitiv yoki xulq-atvor belgilari kamdan-kam hollarda tashxis qo'yilgan birinchi alomatlardir; ular odatda faqat orqada yoki ular yanada rivojlanganda tan olinadi. Kasallikning qay darajada rivojlanganligini bu yordamida o'lchash mumkin birlashgan Xantington kasalligining reyting shkalasi, bu vosita, xulq-atvor, kognitiv va funktsional baholashga asoslangan umumiy reyting tizimini ta'minlaydi.[50][51] Tibbiy tasvir, kabi kompyuter tomografiyasi (CT) va magnit-rezonans tomografiya (MRI), kasallikning boshida kaudat yadrolarining atrofiyasini ko'rsatishi mumkin, bu o'ngdagi rasmda ko'rsatilgan, ammo bu o'zgarishlar o'z-o'zidan HD diagnostikasi emas. Miya atrofiyasi kasallikning rivojlangan bosqichlarida ko'rish mumkin. Funktsional neyroimaging kabi texnikalar funktsional magnit-rezonans tomografiya (fMRI) va pozitron emissiya tomografiyasi (PET), jismoniy simptomlar paydo bo'lishidan oldin miya faoliyatidagi o'zgarishlarni ko'rsatishi mumkin, ammo ular eksperimental vositalar bo'lib, klinik jihatdan qo'llanilmaydi.[19]
Bashoratli genetik test
HD merosxo'rlikning avtosomal dominant modeliga amal qilganligi sababli, uni meros qilib olish xavfi ostida bo'lgan shaxslar uchun tashxis qo'yish uchun kuchli turtki mavjud. The genetik test chunki HD a dan iborat qon testi har birida CAG takrorlanish sonlarini hisoblaydigan HTT allellar.[52] Qisqartirish quyidagicha berilgan:
- 40 yoki undan ortiq CAG takrorlanadi: to'liq penetratsiya allel (FPA).[53] A "ijobiy sinov "yoki" ijobiy natija "odatda ushbu holatga ishora qiladi. Ijobiy natija tashxis deb hisoblanmaydi, chunki uni alomatlar paydo bo'lishidan o'nlab yillar oldin olish mumkin. Ammo, salbiy test bu shaxs genning kengaytirilgan nusxasini olib yurmasligini anglatadi. va HD rivojlanmaydi.[19] Sinov dastlab kasallikni meros qilib olish ehtimoli 50 foiz bo'lgan odamga uning xavfi 100 foizgacha ko'tarilsa yoki yo'q qilinishini aytadi. Kasallik ijobiy natija bergan odam, kasallik paydo bo'lishi uchun etarlicha uzoq umr ko'rishi sharti bilan, hayoti davomida HD rivojlanadi.[19]
- 36 dan 39 gacha takrorlanadi: to'liqsiz yoki kamaytirilgan penetran alleli (RPA). Odatda kattalar hayotida bu alomatlarni keltirib chiqarishi mumkin.[53] RPA bo'lgan odamning 65 yoshida simptomatik bo'lishining maksimal 60% xavfi va 75 yoshida 70% simptomatik bo'lish xavfi mavjud.[53]
- 27 dan 35 gacha takrorlanadi: oraliq allel (IA) yoki katta normal allel. Bu tekshirilgan odamda simptomatik kasallik bilan bog'liq emas, lekin naslga alomatlar berish uchun merosxo'rlik paytida kengayishi mumkin.[53]
- 26 yoki undan kam takrorlash: HD bilan bog'liq emas.[53]
Alomatlar paydo bo'lishidan oldin test o'tkazish hayotni o'zgartiradigan voqea va juda shaxsiy qaror.[19] HD uchun testni tanlashning asosiy sababi mansab va oilaviy qarorlar qabul qilishda yordam berishdir.[19] 1993 yilgacha shaxslar Xantington genini olib yurishini bilish uchun mavjud bo'lmagan. O'sha paytda o'tkazilgan so'rovnomalar shuni ko'rsatdiki, xavf ostida bo'lgan odamlarning 50-70% sinovdan o'tishga qiziqish bildirgan bo'lar edi, ammo bashoratli testlar taklif qilinganligi sababli sinovdan o'tishni tanlaganlar soni ancha kam edi.[54] HDni meros qilib olish xavfi ostida bo'lgan odamlarning 95% dan ko'prog'i, davolanish yo'qligi sababli sinovlarni davom ettirmaydi.[19] Asosiy masala - bu ijobiy natijaning ta'siri bilan taqqoslaganda, odam oxir-oqibat HD rivojlanib borishini bilmaslikdan xavotirda.[19] Natija qanday bo'lishidan qat'i nazar, sinovdan o'tganidan ikki yil o'tgach, stress darajasi pastroq ekanligi aniqlandi, ammo ijobiy natijalar natijasida o'z joniga qasd qilish xavfi ortdi.[19] Buzuqlikni meros qilib olmagan shaxslar duch kelishi mumkin tirik qolganning aybi ta'sirlangan oila a'zolariga nisbatan.[19] Sinovlarni ko'rib chiqishda hisobga olinadigan boshqa omillar qatoriga diskriminatsiya ehtimoli va ijobiy natijaning natijalari kiradi, bu odatda ota-onada ta'sirlangan gen mavjudligini va shaxsning aka-ukalari uni meros qilib olish xavfi borligini anglatadi.[19] Bir tadqiqotda Xantington kasalligi xavfi ostida bo'lgan odamlarning 46 foizida genetik diskriminatsiya aniqlandi. Bu shaxsiy munosabatlar doirasida tibbiy sug'urta yoki mehnat munosabatlariga nisbatan yuqori stavkalarda sodir bo'lgan.[55] Genetik maslahat HD da dastlabki qaror qabul qilish uchun ma'lumot, maslahat va yordam berilishi mumkin, so'ngra tanlangan taqdirda test jarayonining barcha bosqichlarida.[56] Ushbu test natijalari sababli, testdan o'tishni istagan bemorlar uchta konsultatsiya mashg'ulotlarini bajarishlari kerak, ular Xantington haqidagi ma'lumot beradi.[57]
HD uchun genetik tekshiruvdan foydalanish bo'yicha maslahatlar va ko'rsatmalar autosomal dominant kabi boshqa genetik kasalliklar uchun namuna bo'ldi. serebellar ataksiya.[19][58][59] Presemptomatik test HD uchun genetik variantlar bilan boshqa kasalliklarni tekshirishga ham ta'sir ko'rsatdi polikistik buyrak kasallik, oilaviy Altsgeymer kasalligi va ko'krak bezi saratoni.[58] Evropa Molekulyar Genetika Sifati Tarmog'i ushbu kasallik uchun molekulyar genetik tekshiruv uchun har yili tashqi sifatni baholash sxemasini nashr etdi va HD natijalarini sinash va hisobot berishda yordam berish uchun genetik tekshiruv bo'yicha eng yaxshi qo'llanmalarni ishlab chiqdi.[60]
Preimplantatsiya genetik diagnostikasi
Embrionlar yordamida ishlab chiqarilgan ekstrakorporal urug'lantirish HD yordamida genetik tekshiruvdan o'tishi mumkin preimplantatsiya genetik diagnostikasi (PGD). Bir yoki ikkita hujayradan odatda 4-8 hujayrali embriondan ajratib olinadigan va keyinchalik genetik anormallik uchun sinovdan o'tkaziladigan ushbu usul keyinchalik HD genlari ta'sirlangan embrionlarning joylashtirilmasligini ta'minlash uchun ishlatilishi mumkin va shuning uchun har qanday nasl meros qilib olinmaydi. kasallik. Preimplantatsiya genetik diagnostikasining ayrim shakllari - oshkor qilmaslik yoki chetlatish testi - xavf ostida bo'lgan odamlarda HD-bepul naslga ega bo'lishga imkon beradi. holda o'zlarining ota-onalari genotiplarini ochib berishadi, ular HD-ni rivojlantirishga tayyor ekanliklari haqida hech qanday ma'lumot bermaydilar. Chetlatish testida, ta'sirlangan bobo va buvidan HD genini o'z ichiga olgan xromosoma mintaqasini meros qilib olishdan saqlanish uchun embrionlarning DNKsi ota-onalar va bobolari bilan taqqoslanadi. Ma'lumotni oshkor qilmaslik testida bachadondagi faqat kasalliksiz embrionlar almashtiriladi, ota-onalarning genotipi va shu sababli HD uchun ota-onalarning xavfi hech qachon oshkor etilmaydi.[61][62]
Prenatal test
Bundan tashqari, a ni olish mumkin prenatal tashxis embrion uchun yoki homila bachadonda, orqali olingan xomilalik genetik materialdan foydalangan holda chorionik villusdan namuna olish. An amniyosentez homiladorlik 14-18 hafta ichida davom etadigan bo'lsa amalga oshirilishi mumkin. Ushbu protsedura HD mutatsiyasining ko'rsatkichlari uchun chaqaloqni o'rab turgan amniotik suyuqlikni ko'rib chiqadi.[63] Bu ham ota-onalarning genotipini oshkor qilmaslik uchun istisno testi bilan birlashtirilishi mumkin. Prenatal test ota-onada HD tashxisi qo'yilganida, HTT genining kengayishini ko'rsatadigan genetik tekshiruvda bo'lganida yoki kasallikni meros qilib olish ehtimoli 50% bo'lganida amalga oshirilishi mumkin. Ota-onalarga o'zlarining imkoniyatlari haqida maslahat berilishi mumkin, shu jumladan homiladorlikni to'xtatish va aniqlangan genga ega bo'lgan bolaning qiyinchiliklari to'g'risida.[64][65]
Bundan tashqari, ta'sirlangan erkak sherik tufayli xavf ostida bo'lgan homiladorlik paytida, invaziv bo'lmagan prenatal tashxisni tahlil qilish orqali amalga oshirish mumkin hujayrasiz homilaning DNKsi onadan olingan qon namunasida (orqali venipunktur ) homiladorlikning olti va o'n ikki haftalari orasida.[53] Bola tushish xavfi yo'q[53]
Differentsial diagnostika
Oddiy alomatlarga asoslangan HD diagnostikasining taxminan 99% va oila tarixi Kasallikning genetik tekshiruvi HDni keltirib chiqaradigan kengaytirilgan trinukleotid takrorlanishiga ega ekanligini tasdiqlaydi. Qolganlarning aksariyati chaqiriladi HD ga o'xshash (HDL) sindromlar.[19][66] Ko'pgina HDL kasalliklarining sababi noma'lum, ammo ma'lum sabablari bo'lganlar mutatsiyalarga bog'liq prion oqsil geni (HDL1), junktofilin 3 geni (HDL2), retsessiv ravishda meros bo'lib o'tgan noma'lum gen (HDL3 - faqat ikkita oilada uchraydi va yaxshi tushunilmagan) va genni kodlovchi TATA qutisini bog'laydigan oqsil (SCA17, ba'zan HDL4 deb nomlanadi ). HD kabi noto'g'ri tashxis qo'yilishi mumkin bo'lgan boshqa autosomal dominant kasalliklar dentatorubral-pallidoluysian atrofiyasi va neyroferritinopatiya. Shuningdek, bor autosomal retsessiv HD ning sporadik holatlariga o'xshash buzilishlar. Bunga quyidagilar kiradi xorea akantotsitoz va pantotenat kinaz bilan bog'liq neyrodejeneratsiya. Bittasi X bilan bog'langan ushbu turdagi buzuqlik McLeod sindromi.[66]
Menejment
HD-ni davolash mumkin emas, ammo uning ba'zi belgilarining og'irligini kamaytirish uchun davolash usullari mavjud.[67] Ushbu muolajalarning aksariyati uchun HD belgilarini davolashda ularning samaradorligini tasdiqlovchi dalillar to'liq emas.[19][68] Kasallik o'sib borishi bilan o'zingizga g'amxo'rlik qilish qobiliyati pasayadi va ehtiyotkorlik bilan boshqariladi ko'p tarmoqli parvarish qilish tobora zarur bo'lib qolmoqda.[19] Garchi yordam beradigan mashqlar va davolash usullarini nisbatan kam o'rganishgan bo'lsa ham reabilitatsiya qilish HD ning kognitiv alomatlari, foydaliligiga ba'zi dalillar mavjud fizioterapiya, kasbiy terapiya va nutq terapiyasi.[19]
Terapiya
Og'irlikni yo'qotish va ovqatlanishdagi muammolar tufayli yutish qiyinchiliklari va boshqa mushaklarning kelishmovchiligi keng tarqalgan bo'lib, kasallik rivojlanib borishi bilan ovqatlanishni boshqarish tobora muhim ahamiyat kasb etmoqda.[19] Qalinlashtiruvchi vositalar suyuqlikka qo'shilishi mumkin, chunki quyuqroq suyuqlik yutish osonroq va xavfsizroq.[19] Ta'sirlangan odamga asta-sekin ovqat eyishni va og'ziga kichikroq ovqat olib borishni eslatish ham bo'g'ilishning oldini olish uchun ishlatilishi mumkin.[19] Agar ovqatlanish juda xavfli yoki noqulay bo'lib qolsa, a dan foydalanish imkoniyati teri osti endoskopik gastrostomiya mavjud. Bu doimiy ravishda biriktirilgan oziqlantiruvchi naycha qorin ichiga oshqozon, bu esa xavfni kamaytiradi intiluvchan oziq-ovqat va ovqatlanishni yaxshiroq boshqarishni ta'minlaydi.[69] Tomonidan baholash va boshqarish logoped-patologlar Xantington kasalligi bo'yicha tajribaga ega bo'lish tavsiya etiladi.[19]
Xantington kasalligi bo'lgan odamlar a kasalligini ko'rishlari mumkin fizioterapevt jismoniy simptomlarni davolashning invaziv bo'lmagan va dorilarga asoslangan bo'lmagan usullari uchun. Fizik terapevtlar tushish xavfini baholash va oldini olish, shuningdek kuchaytirish, cho'zish va yurak-qon tomir mashqlarini amalga oshirishi mumkin. Yurish uchun yordamchi vositalar tegishli ravishda tayinlanishi mumkin. Jismoniy terapevtlar shuningdek, nafas olish mashqlarini va havo yo'llarini tozalash texnikasi nafas olish muammolarini rivojlanishi bilan.[70] Xantington kasalligida fizioterapiya bo'yicha konsensus ko'rsatmalari Evropa HD tarmog'i.[70] Erta maqsadlar reabilitatsiya aralashuvlar funktsiya yo'qolishining oldini olishdir. Kasallikning dastlabki va o'rta bosqichlarida reabilitatsiya dasturlarida ishtirok etish foydali bo'lishi mumkin, chunki bu vosita va funktsional ko'rsatkichlarni uzoq muddatli saqlashga aylanadi. Kechki bosqichda reabilitatsiya vosita va funktsional yo'qotishlarni qoplashga qaratilgan.[71] Uzoq muddatli mustaqil boshqarish uchun terapevt tegishli odamlar uchun uyda mashq qilish dasturlarini ishlab chiqishi mumkin.[72]
Bundan tashqari, Xantington kasalligiga chalinganlarning soni tobora ko'payib bormoqda, boshqa davolash usullaridan tashqari og'ir kasallik alomatlari va stressini davolash orqali hayot sifatini yaxshilashga qaratilgan palyatif yordamga murojaat qilishadi.[73]
Dori vositalari
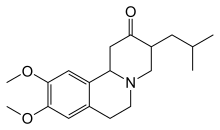
Tetrabenazin 2000 yilda Evropa Ittifoqida Xantington kasalligida xoreyani davolash uchun va 2008 yilda AQShda tasdiqlangan.[74] Xoreyani kamaytirishga yordam beradigan boshqa dorilar kiradi antipsikotiklar va benzodiazepinlar.[15] Kabi birikmalar amantadin yoki remasemid hanuzgacha tergov qilinmoqda, ammo dastlabki ijobiy natijalarni ko'rsatmoqda.[19] Gipokinesiya va qat'iylik, ayniqsa, voyaga etmaganlar ishida davolash mumkin antiparkinsoniyalik giyohvand moddalar va miyoklonik giperkineziya bilan davolash mumkin valproik kislota.[15] Taxminiy dalillar topildi etil eikosapentaenoik kislota vosita simptomlarini bir yilda yaxshilash.[75] 2017 yilda Deutetrabenazin HD-da xoreyani davolash uchun tetrabenazin dori-darmonlarining og'ir shakli FDA tomonidan tasdiqlangan.[76] Bu shunday sotiladi Ostedo va birinchi kichik molekulali dori FDA roziligini olish.[77]
Psixiatrik alomatlarni umumiy populyatsiyada ishlatiladigan dorilarga o'xshash davolash mumkin.[19][68] Selektiv serotoninni qaytarib olish inhibitörleri va mirtazapin depressiya uchun tavsiya etilgan, ammo atipik antipsikotiklar uchun tavsiya etiladi psixoz va yurish-turish muammolari.[68] Odamlar bir nechta dori-darmonlarni birgalikda davolash bilan uzoq muddatli davolanishni talab qilishi mumkinligi sababli, mutaxassisga neyropsikiyatrik kirish tavsiya etiladi.[19]
Ta'lim
Jismoniy shaxslarning oilalari va jamiyat umuman olganda, HD-ni meros qilib olgan yoki merosxo'rlik xavfi ostida bo'lganlar, HD-ning avlodlari tajribasiga ega, ammo kasallikni tushunishda so'nggi yutuqlar va genetik testlarning mavjudligini bilmasligi mumkin. Genetik maslahat bilimlarini yangilash, ulardagi har qanday asossiz e'tiqodlarni yo'q qilish va kelajakdagi imkoniyatlari va rejalarini ko'rib chiqishda yordam berish orqali ushbu shaxslarga foyda keltiradi. Shuningdek, oilani rejalashtirishni tanlash, parvarish qilishni boshqarish va boshqa masalalarga tegishli ma'lumotlar keltirilgan.[19][78]
Prognoz
Trinukleotid takrorlanishining davomiyligi semptomlar paydo bo'lish yoshi va ularning rivojlanish tezligining 60% ini tashkil qiladi. Keyinchalik takrorlash erta boshlanish yoshiga va simptomlarning tezroq rivojlanishiga olib keladi.[19][79] Oltmishdan ortiq takroriy kasallikka chalingan shaxslar ko'pincha 20 yoshgacha kasallikni rivojlantiradi, 40 dan kam takrorlanadiganlar esa asemptomatik bo'lib qolishi mumkin.[80] The remaining variation is due to environmental factors and other genes that influence the mechanism of the disease.[19]
Life expectancy in HD is generally around 20 years following the onset of visible symptoms.[19] Most life-threatening complications result from muscle coordination and, to a lesser extent, behavioral changes induced by declining cognitive function. The largest risk is zotiljam, which causes death in one third of those with HD. As the ability to synchronize movements deteriorates, difficulty clearing the lungs and an increased risk of intiluvchan food or drink both increase the risk of contracting pneumonia. The second greatest risk is yurak kasalligi, which causes almost a quarter of fatalities of those with HD.[19] O'z joniga qasd qilish is the third greatest cause of fatalities, with 7.3% of those with HD taking their own lives and up to 27% attempting to do so. It is unclear to what extent suicidal thoughts are influenced by behavioral symptoms, as they signify sufferers' desires to avoid the later stages of the disease.[81][82][83] Other associated risks include choking, physical injury from falls, and malnutrition.[19]
Epidemiologiya
The late onset of Huntington's disease means it does not usually affect reproduction.[19] Dunyo bo'ylab tarqalishi of HD is 5–10 cases per 100,000 persons,[84][85] but varies greatly geographically as a result of ethnicity, local migration and past immigration patterns.[19] Prevalence is similar for men and women. The rate of occurrence is highest in xalqlar of Western European descent, averaging around 7 per 100,000 people, and is lower in the rest of the world; e.g., one per million people of Asian and African descent. A 2013 epidemiological study of the prevalence of Huntington's disease in the UK between 1990 and 2010 found that the average prevalence for the UK was 12.3 per 100,000.[19][86] Additionally, some localized areas have a much higher prevalence than their regional average.[19] One of the highest incidences is in the isolated populations of the Marakaybo ko'li viloyati Venesuela, where HD affects up to 700 per 100,000 persons.[19][87] Other areas of high localization have been found in Tasmaniya and specific regions of Shotlandiya, Uels va Shvetsiya.[83] Increased prevalence in some cases occurs due to a local asoschining ta'siri, a historical migration of carriers into an area of geografik izolyatsiya.[83][88] Some of these carriers have been traced back hundreds of years using nasabga oid tadqiqotlar.[83] Genetik haplotiplar can also give clues for the geographic variations of prevalence.[83][89] Islandiya, on the contrary, has a rather low prevalence of 1 per 100,000, despite the fact that Islandiyaliklar as a people are descended of the early Germanic tribes of Scandinavia which also gave rise to the Shvedlar; all cases with the exception of one going back nearly two centuries having derived from the offspring of a couple living early in the 19th century.[90] Finlyandiya, as well, has a low incidence of only 2.2 per 100,000 people.[91]
Until the discovery of a genetik test, statistics could only include klinik diagnostika based on physical symptoms and a oila tarixi of HD, excluding those who died of other causes before diagnosis. These cases can now be included in statistics; and, as the test becomes more widely available, estimates of the prevalence and incidence of the disorder are likely to increase.[83][92]
Tarix

Although Huntington's has been recognized as a disorder since at least the O'rta yosh, the cause has been unknown until fairly recently. Huntington's was given different names throughout this history as understanding of the disease changed. Originally called simply 'chorea' for the jerky dancelike movements associated with the disease, HD has also been called "hereditary chorea" and "chronic progressive chorea".[94] The first definite mention of HD was in a letter by Charles Oscar Waters, published in the first edition of Robli Dunglison "s Tibbiyot amaliyoti in 1842. Waters described "a form of chorea, vulgarly called magrums", including accurate descriptions of the chorea, its progression, and the strong heredity of the disease.[95] 1846 yilda Charlz Gorman observed how higher prevalence seemed to occur in localized regions.[95] Independently of Gorman and Waters, both students of Dunglison at Jefferson tibbiyot kolleji Filadelfiyada,[96] Johan Christian Lund also produced an early description in 1860.[95] He specifically noted that in Setesdalen, a secluded mountain valley in Norvegiya, there was a high prevalence of dementia associated with a pattern of jerking movement disorders that ran in families.[97]
The first thorough description of the disease was by George Huntington in 1872. Examining the combined medical history of several generations of a family exhibiting similar symptoms, he realized their conditions must be linked; he presented his detailed and accurate definition of the disease as his first paper. Huntington described the exact pattern of inheritance of autosomal dominant disease years before the rediscovery by scientists of Mendeliyalik meros.
Of its hereditary nature. When either or both the parents have shown manifestations of the disease ... one or more of the offspring almost invariably suffer from the disease ... But if by any chance these children go through life without it, the thread is broken and the grandchildren and great-grandchildren of the original shakers may rest assured that they are free from the disease.[93][98]
Janob Uilyam Osler was interested in the disorder and chorea in general, and was impressed with Huntington's paper, stating that "In the history of medicine, there are few instances in which a disease has been more accurately, more graphically or more briefly described."[95][99] Osler's continued interest in HD, combined with his influence in the field of medicine, helped to rapidly spread awareness and knowledge of the disorder throughout the medical community.[95] Great interest was shown by scientists in Europe, including Louis Théophile Joseph Landouzy, Désiré-Magloire Bourneville, Camillo Golgi va Jozef Jyul Dejerin, and until the end of the century, much of the research into HD was European in origin.[95] By the end of the 19th century, research and reports on HD had been published in many countries and the disease was recognized as a worldwide condition.[95]
During the rediscovery of Mendelian inheritance at the turn of the 20th century, HD was used tentatively as an example of autosomal dominant inheritance.[95] The English biologist Uilyam Bateson used the pedigrees of affected families to establish that HD had an autosomal dominant inheritance pattern.[96] The strong inheritance pattern prompted several researchers, including Smit Ely Jelliff, to attempt to trace and connect family members of previous studies.[95] Jelliffe collected information from across Nyu York and published several articles regarding the genealogy of HD in Yangi Angliya.[100] Jelliffe's research roused the interest of his college friend, Charlz Davenport, who commissioned Elizabeth Muncey to produce the first field study on the AQShning Sharqiy qirg'og'i of families with HD and to construct their pedigrees.[101] Davenport used this information to document the variable age of onset and range of symptoms of HD; he claimed that most cases of HD in the USA could be traced back to a handful of individuals.[101] This research was further embellished in 1932 by P. R. Vessie, who popularized the idea that three brothers who left Angliya in 1630 bound for Boston were the progenitors of HD in the USA.[102] The claim that the earliest progenitors had been established and evgenik bias of Muncey's, Davenport's, and Vessie's work contributed to misunderstandings and prejudice about HD.[96] Muncey and Davenport also popularized the idea that in the past some HD sufferers may have been thought to be possessed by spirits or victims of sehrgarlik, and were sometimes chetlandi yoki surgun qilingan by society.[103][104] This idea has not been proven. Researchers have found contrary evidence; for instance, the community of the family studied by George Huntington openly accommodated those who exhibited symptoms of HD.[96][103]
The search for the cause of this condition was enhanced considerably in 1968, when the Herediter Disease Foundation (HDF) was created by Milton Veksler, a psixoanalist asoslangan Los Anjeles, Kaliforniya, whose wife Leonore Sabin had been diagnosed earlier that year with Huntington's disease.[105] The three brothers of Wexler's wife also suffered from this disease.
The foundation was involved in the recruitment of more than 100 scientists in the US-Venezuela Huntington's Disease Collaborative Project who over a 10-year period from 1979, worked to locate the genetic cause.[106] This was achieved in 1983 when a causal gene was approximately located,[88] and in 1993 the gene was precisely located at chromosome 4 (4p16.3).[107] The study had focused on the populations of two isolated Venesuela villages, Barranquitas and Lagunetas, where there was an unusually high prevalence of the disease. It involved over 18,000 people, mostly from a single extended family, and resulted in making HD the first autosomal kasallik lokus found using genetic linkage analysis.[107][108] Among other innovations, the project developed DNA-marking methods which were an important step in making the Inson genomining loyihasi mumkin.[106]
In the same time frame, key discoveries concerning the mechanisms of the disorder were being made, including the findings by Anita Harding 's research group on the effects of the gene's length.[109]
Modelling the disease in various types of animals, such as the transgenik mouse developed in 1996, enabled larger scale experiments. As these animals have faster metabolizm and much shorter lifespans than humans, results from experiments are received sooner, speeding research. The 1997 discovery that mhtt fragments misfold led to the discovery of the nuclear inclusions they cause. These advances have led to increasingly extensive research into the proteins involved with the disease, potential drug treatments, care methods, and the gene itself.[95][110]
The condition was formerly called 'Huntington's chorea' but this term has been replaced by 'Huntington's disease' because not all patients develop chorea and due to the importance of cognitive and behavioral problems.[111]
Jamiyat va madaniyat
Axloq qoidalari
Huntington's disease, particularly the application of the genetic test for the disease, has raised several ethical issues. The issues for genetic testing include defining how mature an individual should be before being considered eligible for testing, ensuring the confidentiality of results, and whether companies should be allowed to use test results for decisions on employment, life insurance or other financial matters. There was controversy when Charlz Davenport proposed in 1910 that majburiy sterilizatsiya va immigratsiya control be used for people with certain diseases, including HD, as part of the evgenika harakat.[112] In vitro urug'lantirish has some issues regarding its use of embryos. Some HD research has ethical issues due to its use of hayvonlarni sinovdan o'tkazish va embrional ildiz hujayralari.[113][114]
The development of an accurate diagnostic test for Huntington's disease has caused social, legal, and ethical concerns over access to and use of a person's results.[115][116]Many guidelines and testing procedures have strict procedures for disclosure and confidentiality to allow individuals to decide when and how to receive their results and also to whom the results are made available.[19] Moliya institutlari and businesses are faced with the question of whether to use genetic test results when assessing an individual, such as for life insurance or employment. The United Kingdom's insurance companies agreed with the Sog'liqni saqlash va ijtimoiy yordam bo'limi that until 2017 customers would not need to disclose predictive genetics tests to them, but this agreement explicitly excluded the government-approved test for Huntington's when writing policies with a value over GB£500,000.[117][118] As with other untreatable genetic conditions with a later onset, it is ethically questionable to perform pre-symptomatic testing on a child or adolescent, as there would be no medical benefit for that individual. There is consensus for testing only individuals who are considered cognitively mature, although there is a counter-argument that parents have a right to make the decision on their child's behalf. With the lack of an effective treatment, testing a person under qonuniy yosh who is not judged to be vakolatli is considered unethical in most cases.[39][119][120]
There are ethical concerns related to prenatal genetic testing yoki preimplantatsiya genetik diagnostikasi to ensure a child is not born with a given disease.[121] For example, prenatal testing raises the issue of selective abortion, a choice considered unacceptable by some.[121] As it is a dominant disease, there are difficulties in situations in which a parent does not want to know his or her own diagnosis. This would require parts of the process to be kept secret from the parent.[121]
Support organizations

In 1968, after experiencing HD in his wife's family, Dr. Milton Wexler was inspired to start the Herediter Disease Foundation (HDF), with the aim of curing genetic illnesses by coordinating and supporting research.[11] The foundation and Wexler's daughter, Nensi Veksler, were key parts of the research team in Venezuela which discovered the HD gene.[11]
At roughly the same time as the HDF formed, Marjori Gutri helped to found the Committee to Combat Huntington's Disease (now the Amerikaning Xantington kasalliklari jamiyati ), eridan keyin Vudi Gutri died from complications of HD.[12]
Since then, support and research organizations have formed in many countries around the world and have helped to increase public awareness of HD. A number of these collaborate in umbrella organizations, like the International Huntington Association and the European HD network.[122] Many support organizations hold an annual HD awareness event, some of which have been endorsed by their respective governments. For example, 6 June is designated "National Huntington's Disease Awareness Day" by the AQSh Senati.[123]
The largest funder of Huntington's disease research globally,[124] bo'ladi Cure Huntington's Disease Initiative Foundation (CHDI), a US foyda keltirmaydigan biomedical foundation that aims to "rapidly discover and develop drugs that delay or slow Huntington's disease".[125] CHDI was formerly known as the High Q Foundation. In 2006, it spent $50 million on Huntington's disease research.[124] CHDI collaborates with many academic and commercial laboratories globally and engages in oversight and management of research projects as well as funding.[126] Many organizations exist to support and inform those affected by HD, including the Huntington's Disease Association Buyuk Britaniyada.
Tadqiqot yo'nalishlari
Research into the mechanism of HD is focused on identifying the functioning of HTT, how mhtt differs or interferes with it, and the brain pathology that the disease produces.[127] Research is conducted using in vitro methods, animal models and human volunteers. Animal models are critical for understanding the fundamental mechanisms causing the disease and for supporting the early stages of giyohvand moddalarni ishlab chiqarish.[110] Animals with chemically induced brain injury exhibit HD-like symptoms and were initially used, but they did not mimic the progressive features of the disease.[128] The identification of the causative gene has enabled the development of many transgenic animal modellari, shu jumladan nematod qurtlar, Drosophila mevali chivinlar, mice, rats, sheep, pigs and monkeys that express mutant huntingtin and develop progressive neyrodejeneratsiya and HD-like symptoms.[110]
Research is being conducted on many different approaches to prevent Huntington's disease or slow its progression.[127] Disease-modifying strategies can be broadly grouped into three categories: reducing the level of the mutant huntingtin protein (including gene splicing va genlarni susaytirish ); approaches aimed at improving neuronal survival by reducing the harm caused by the protein to specific cellular pathways and mechanisms (including protein homeostasis va giston deatsetilaza inhibition); and strategies to replace lost neurons. In addition, novel therapies to improve brain functioning are under development; these seek to produce symptomatic rather than disease-modifying therapies, and include fosfodiesteraza inhibitörleri.[129][130]
In 2020 the CHDI Foundation began a small-molecule computational research collaboration with OpenEye Scientific focusing on small-molecule treatments, using a molecular design platform of OpenEye's known as Orion.[125]
Reducing huntingtin production
Genlarning susayishi aims to reduce the production of the mutant protein, since HD is caused by a single dominant gene encoding a toxic protein. Gene silencing experiments in mouse models have shown that when the expression of mhtt is reduced, symptoms improve.[131] The safety of RNK aralashuvi va allele-specific oligonucleotide (ASO) methods of gene silencing has been demonstrated in mice and the larger primate macaque brain.[132][133] Allele-specific silencing attempts to silence mutant htt while leaving wild-type HTT untouched. One way of accomplishing this is to identify polymorphisms present on only one allele and produce gene silencing drugs that target polymorphisms in only the mutant allele.[134] The first gene silencing trial involving humans with HD began in 2015, testing the safety of IONIS-HTTRx, produced by Ionis farmatsevtika va boshchiligida UCL Nevrologiya instituti.[135][136] Mutant huntingtin was detected and quantified for the first time in miya omurilik suyuqligi from Huntington's disease mutation-carriers in 2015 using a novel "single-molecule counting" immunoassay,[137] providing a direct way to assess whether huntingtin-lowering treatments are achieving the desired effect.[138][139] Xuddi shunday, gene splicing techniques are being looked at to try to repair a genome with the erroneous gene that causes HD, using tools such as CRISPR / Cas9.[130]
Increasing huntingtin clearance
Another strategy to reduce the levels of mutant huntingtin is to increase the rate at which cells are able to clear the mutant protein.[140] As mutant huntingtin protein (and many other aggregate prone proteins) is degraded by autophagy, increasing levels of autophagy have the potential to reduce levels of the toxic protein and thereby ameliorate disease.[141] Pharmacological and genetic inducers of autophagy have been tested in a variety of Huntington's disease models, and many have been shown to reduce mHTT levels and decrease toxicity.[140]
Improving cell survival
Among the approaches aimed at improving cell survival in the presence of mutant huntingtin are correction of transkripsiyani tartibga solish foydalanish giston deatsetilaza inhibitörleri, modulyatsiya birlashma of huntingtin, improving metabolizm va mitoxondriyal funktsiya and restoring function of sinapslar.[131]
Neuronal replacement
Ildiz hujayralari terapiyasi is the replacement of damaged neurons by transplantation of ildiz hujayralari into affected regions of the brain. Experiments have yielded mixed results using this technique in animal models and preliminary human klinik sinovlar.[142] Whatever their future therapeutic potential, stem cells are already a valuable tool for studying Huntington's disease in the laboratory.[143]
Klinik sinovlar
In 2020 there were 197 klinik sinovlar related to varied therapies and biomarkers for Huntington's disease listed as either underway, recruiting or newly completed.[144]
Murakkab moddalar sudlangan, that have failed to prevent or slow the progression of Huntington's disease include remacemide, koenzim Q10, riluzol, kreatin, minosiklin, ethyl-EPA, fenilbutirat va dimebon.[145]
Shuningdek qarang
 Tibbiyot portali
Tibbiyot portali
Adabiyotlar
- ^ a b v d e f g h men j k l Dayalu P, Albin RL (February 2015). "Huntington disease: pathogenesis and treatment". Nevrologik klinikalar. 33 (1): 101–14. doi:10.1016/j.ncl.2014.09.003. PMID 25432725.
- ^ a b v d e f g Caron NS, Wright GE, Hayden MR (2020). Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Stephens K, Amemiya A (eds.). "Huntington Disease". GeneReviews. PMID 20301482.
- ^ a b v d e f g h men j k l Frank S (January 2014). "Treatment of Huntington's disease". Neyroterapevtikalar. 11 (1): 153–60. doi:10.1007/s13311-013-0244-z. PMC 3899480. PMID 24366610.
- ^ a b v d e f g h "Huntington's Disease Information Page | National Institute of Neurological Disorders and Stroke". www.ninds.nih.gov. Olingan 14 dekabr 2020.
- ^ a b Durr A, Gargiulo M, Feingold J (November 2012). "The presymptomatic phase of Huntington disease". Revue Neurologique. 168 (11): 806–8. doi:10.1016/j.neurol.2012.07.003. PMID 22902173.
- ^ Ferri, Fred F. (2010). Ferri's differential diagnosis : a practical guide to the differential diagnosis of symptoms, signs, and clinical disorders (2-nashr). Philadelphia, PA: Elsevier/Mosby. p. Chapter H. ISBN 978-0323076999.
- ^ a b "Molecular Pathogenesis in Huntington's Disease". protein.bio.msu.ru. Olingan 8 noyabr 2020.
- ^ a b v d e Bates GP, Dorsey R, Gusella JF, Hayden MR, Kay C, Leavitt BR, Nance M, Ross CA, Scahill RI, Wetzel R, Wild EJ, Tabrizi SJ (2015 yil aprel). "Huntington disease". Tabiat sharhlari kasalliklarga qarshi asarlar. 1: 15005. doi:10.1038/nrdp.2015.5. PMID 27188817. S2CID 25759303.
- ^ a b Vale TC, Cardoso F (2015). "Chorea: A Journey through History". Tremor and Other Hyperkinetic Movements. 5. doi:10.7916/D8WM1C98. PMC 4454991. PMID 26056609.
- ^ a b "Learning About Huntington's Disease". www.genome.gov. Arxivlandi asl nusxasidan 2016 yil 4 iyulda. Olingan 19 iyul 2016.
- ^ a b v d "History of the HDF". Hereditary Disease Foundation. Arxivlandi asl nusxasi 2015 yil 19-noyabrda. Olingan 18 noyabr 2015.
- ^ a b "History and Genetics of Huntington's Disease | Huntington's Disease Society of America". Olingan 14 dekabr 2020.
- ^ a b v d van Duijn E, Kingma EM, van der Mast RC (2007). "Psychopathology in verified Huntington's disease gene carriers". Nöropsikiyatri va klinik nevrologiya jurnali. 19 (4): 441–8. doi:10.1176/appi.neuropsych.19.4.441. PMID 18070848.
- ^ "Xantington kasalligi". www.nhsinform.scot. Olingan 12 iyul 2020.
- ^ a b v "Xantington kasalligi". genereviews bookshelf. Vashington universiteti. Iyun 2020. Olingan 22 noyabr 2020.
- ^ Ruhiy kasalliklar diagnostikasi va statistik qo'llanmasi: DSM-5 (5-nashr). Arlington, VA: Amerika psixiatriya assotsiatsiyasi. 2013. p. 639. ISBN 9780890425541.
- ^ a b Kremer B (2002). "Clinical neurology of Huntington's disease". In Bates G, Harper P, Jones L (eds.). Huntington's Disease – Third Edition. Oksford: Oksford universiteti matbuoti. pp. 28–53. ISBN 978-0-19-851060-4.
- ^ Wagle AC, Wagle SA, Marková IS, Berrios GE (2000). "Psychiatric Morbidity in Huntington's disease". Neurology, Psychiatry and Brain Research (8): 5–16.
- ^ a b v d e f g h men j k l m n o p q r s t siz v w x y z aa ab ak reklama ae af ag ah ai aj ak al am an ao ap aq ar kabi da au av aw bolta ay az ba bb miloddan avvalgi bd bo'lishi bf bg bh bi bj bk bl bm bn bo bp Walker FO (January 2007). "Xantington kasalligi". Lanset. 369 (9557): 218–28. doi:10.1016 / S0140-6736 (07) 60111-1. PMID 17240289. S2CID 46151626.
- ^ a b v d e f g Montoya A, Price BH, Menear M, Lepage M (January 2006). "Brain imaging and cognitive dysfunctions in Huntington's disease" (PDF). Psixiatriya va nevrologiya jurnali. 31 (1): 21–9. PMC 1325063. PMID 16496032. Arxivlandi asl nusxasi (PDF) 2016 yil 23 martda. Olingan 17 sentyabr 2008.
- ^ a b Dickey AS, La Spada AR (April 2018). "Therapy development in Huntington disease: From current strategies to emerging opportunities". Amerika tibbiyot genetikasi jurnali. A qism. 176 (4): 842–861. doi:10.1002/ajmg.a.38494. PMC 5975251. PMID 29218782.
- ^ Aziz NA, van der Marck MA, Pijl H, Olde Rikkert MG, Bloem BR, Roos RA (December 2008). "Weight loss in neurodegenerative disorders". Nevrologiya jurnali. 255 (12): 1872–80. doi:10.1007/s00415-009-0062-8. PMID 19165531. S2CID 26109381.
- ^ "Booklet by the Huntington Society of Canada" (PDF). Caregiver's Handbook for Advanced-Stage Huntington Disease. HD Society of Canada. 11 Aprel 2007. Arxivlangan asl nusxasi (PDF) 2008 yil 25-iyunda. Olingan 10 avgust 2008.
- ^ Murray ED, Buttner N, Narx BH (2012). "Nevrologik amaliyotda depressiya va psixoz". Bredli WG, Daroff RB, Fenichel GM, Yankovich J (tahr.). Bredlining nevrologiyasi klinik amaliyotda (6-nashr). Filadelfiya, Pensilvaniya: Elsevier / Sonders. p. 108. ISBN 978-1-4377-0434-1.
- ^ van der Burg JM, Björkqvist M, Brundin P (August 2009). "Beyond the brain: widespread pathology in Huntington's disease". Lanset. Nevrologiya. 8 (8): 765–74. doi:10.1016/S1474-4422(09)70178-4. PMID 19608102. S2CID 14419437.
- ^ Katsuno M, Banno H, Suzuki K, Takeuchi Y, Kawashima M, Tanaka F, Adachi H, Sobue G (May 2008). "Molecular genetics and biomarkers of polyglutamine diseases". Hozirgi molekulyar tibbiyot. 8 (3): 221–34. doi:10.2174/156652408784221298. PMID 18473821.
- ^ Squitieri F, Frati L, Ciarmiello A, Lastoria S, Quarrell O (February 2006). "Juvenile Huntington's disease: does a dosage-effect pathogenic mechanism differ from the classical adult disease?". Qarish va rivojlanish mexanizmlari. 127 (2): 208–12. doi:10.1016/j.mad.2005.09.012. PMID 16274727. S2CID 20523093.
- ^ Nance MA, Myers RH (2001). "Juvenile onset Huntington's disease--clinical and research perspectives". Aqliy rivojlanishning sustligi va rivojlanishdagi nogironlikning tadqiqotlari. 7 (3): 153–7. doi:10.1002/mrdd.1022. PMID 11553930.
- ^ Passarge E (2001). Color Atlas of Genetics (2-nashr). Thieme. p.142. ISBN 978-0-86577-958-7.
- ^ Ridley RM, Frith CD, Crow TJ, Conneally PM (September 1988). "Anticipation in Huntington's disease is inherited through the male line but may originate in the female". Tibbiy genetika jurnali. 25 (9): 589–95. doi:10.1136/jmg.25.9.589. PMC 1051535. PMID 2972838.
- ^ Semaka A, Creighton S, Warby S, Hayden MR (October 2006). "Predictive testing for Huntington disease: interpretation and significance of intermediate alleles". Klinik genetika. 70 (4): 283–94. doi:10.1111/j.1399-0004.2006.00668.x. PMID 16965319. S2CID 26007984.
- ^ Wexler NS, Young AB, Tanzi RE, Travers H, Starosta-Rubinstein S, Penney JB, Snodgrass SR, Shoulson I, Gomez F, Ramos Arroyo MA (1987). "Homozygotes for Huntington's disease" (PDF). Tabiat. 326 (6109): 194–7. Bibcode:1987Natur.326..194W. doi:10.1038/326194a0. hdl:2027.42/62543. PMID 2881213. S2CID 4312171.
- ^ Squitieri F, Gellera C, Cannella M, Mariotti C, Cislaghi G, Rubinsztein DC, Almqvist EW, Turner D, Bachoud-Lévi AC, Simpson SA, Delatycki M, Maglione V, Hayden MR, Donato SD (April 2003). "Homozygosity for CAG mutation in Huntington disease is associated with a more severe clinical course". Miya. 126 (Pt 4): 946–55. doi:10.1093/brain/awg077. PMID 12615650.
- ^ Goehler H, Lalowski M, Stelzl U, Waelter S, Stroedicke M, Worm U, Droege A, Lindenberg KS, Knoblich M, Haenig C, Herbst M, Suopanki J, Scherzinger E, Abraham C, Bauer B, Hasenbank R, Fritzsche A, Ludewig AH, Büssow K, Buessow K, Coleman SH, Gutekunst CA, Landwehrmeyer BG, Lehrach H, Wanker EE (September 2004). "Hinttin agregatsiyasini kuchaytiruvchi GIT1 bilan oqsillarning o'zaro aloqasi tarmog'i Xantington kasalligi bilan bog'lanadi". Molekulyar hujayra. 15 (6): 853–65. doi:10.1016 / j.molcel.2004.09.016. PMID 15383276.
- ^ Glajch KE, Sadri-Vakili G (2015). "Epigenetic Mechanisms Involved in Huntington's Disease Pathogenesis". Xantington kasalligi jurnali. 4 (1): 1–15. doi:10.3233/JHD-159001. PMID 25813218.
- ^ Harjes P, Wanker EE (August 2003). "The hunt for huntingtin function: interaction partners tell many different stories". Biokimyo fanlari tendentsiyalari. 28 (8): 425–33. doi:10.1016/S0968-0004(03)00168-3. PMID 12932731.
- ^ a b v Cattaneo E, Zuccato C, Tartari M (December 2005). "Normal huntingtin function: an alternative approach to Huntington's disease". Neuroscience-ning tabiat sharhlari. 6 (12): 919–30. doi:10.1038/nrn1806. PMID 16288298. S2CID 10119487.
- ^ a b v d e Rubinsztein DC, Carmichael J (August 2003). "Huntington's disease: molecular basis of neurodegeneration". Molekulyar tibbiyot bo'yicha ekspertlar. 5 (20): 1–21. doi:10.1017/S1462399403006549. PMID 14585171.
- ^ a b Bloch M, Hayden MR (January 1990). "Opinion: predictive testing for Huntington disease in childhood: challenges and implications". Amerika inson genetikasi jurnali. 46 (1): 1–4. PMC 1683548. PMID 2136787.
- ^ a b v Sadri-Vakili G, Cha JH (June 2006). "Mechanisms of disease: Histone modifications in Huntington's disease". Tabiat klinikasi nevrologiyasi. 2 (6): 330–8. doi:10.1038/ncpneuro0199. PMID 16932577. S2CID 12474262.
- ^ Liu Z, Chjou T, Ziegler AC, Dimitrion P, Zuo L (2017). "Neyrodejenerativ kasalliklarda oksidlovchi stress: molekulyar mexanizmlardan klinik qo'llanmalargacha". Oksidlovchi tibbiyot va uyali uzoq umr ko'rish. 2017: 2525967. doi:10.1155/2017/2525967. PMC 5529664. PMID 28785371.
- ^ Kumar A, Ratan RR (October 2016). "Oxidative Stress and Huntington's Disease: The Good, The Bad, and The Ugly". Xantington kasalligi jurnali. 5 (3): 217–237. doi:10.3233/JHD-160205. PMC 5310831. PMID 27662334.
- ^ Purves D, Augustine GA, Fitzpatrick D, Hall W, LaMantia AS, McNamara JO, Williams SM (2001). "Modulation of Movement by the Basal Ganglia – Circuits within the Basal Ganglia System". In Purves D (ed.). Nevrologiya (2-nashr). Sanderlend, MA: Sinauer Associates. ISBN 978-0-87893-742-4. Arxivlandi asl nusxasidan 2009 yil 18 fevralda. Olingan 1 aprel 2009.
- ^ Lobsiger CS, Klivlend DW (2007 yil noyabr). "Glial cells as intrinsic components of non-cell-autonomous neurodegenerative disease". Tabiat nevrologiyasi. 10 (11): 1355–60. doi:10.1038 / nn1988. PMC 3110080. PMID 17965655.
- ^ a b Crossman AR (may 2000). "Harakat buzilishlarining funktsional anatomiyasi". Anatomiya jurnali. 196 ( Pt 4) (4): 519–25. doi:10.1046 / j.1469-7580.2000.19640519.x. PMC 1468094. PMID 10923984.
- ^ Duffy J (2013). Motor Speech Disorders: Substrates, Differential Diagnosis, and Management (3-nashr). Sent-Luis, Missuri: Elsevier. pp. 196–7.
- ^ a b Petruska J, Xartenstine MJ, Goodman MF (1998 yil fevral). "Neyrodejenerativ kasallik bilan bog'liq bo'lgan CAG / CTG triplet takrorlanishining DNK-polimeraza kengayishidagi ip siljishini tahlil qilish". Biologik kimyo jurnali. 273 (9): 5204–10. doi:10.1074 / jbc.273.9.5204. PMID 9478975.
- ^ Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL, et al. (Oktyabr 2001). "Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila". Tabiat. 413 (6857): 739–43. Bibcode:2001Natur.413..739S. doi:10.1038/35099568. PMID 11607033. S2CID 4419980.
- ^ Gaillard F (1 May 2007). "Xantington kasalligi". Radiology picture of the day. www.radpod.org. Arxivlandi asl nusxasi 2007 yil 22 oktyabrda. Olingan 24 iyul 2009.
- ^ Rao AK, Muratori L, Louis ED, Moskowitz CB, Marder KS (April 2009). "Clinical measurement of mobility and balance impairments in Huntington's disease: validity and responsiveness". Yurish va holat. 29 (3): 433–6. doi:10.1016/j.gaitpost.2008.11.002. PMID 19111470.
- ^ "Unified Huntington's Disease Rating Scale (UHDRS)". UHDRS and Database. HSG. 2009 yil 1-fevral. Arxivlandi asl nusxasidan 2015 yil 11 avgustda. Olingan 14 aprel 2009.
- ^ Myers RH (April 2004). "Huntington's disease genetics". NeuroRx. 1 (2): 255–62. doi:10.1602/neurorx.1.2.255. PMC 534940. PMID 15717026.
- ^ a b v d e f g de Die-Smulders CE, de Wert GM, Liebaers I, Tibben A, Evers-Kiebooms G (May 2013). "Reproductive options for prospective parents in families with Huntington's disease: clinical, psychological and ethical reflections". Inson ko'payishining yangilanishi. 19 (3): 304–15. doi:10.1093/humupd/dms058. PMID 23377865. de Die-Smulders CE, de Wert GM, Liebaers I, Tibben A, Evers-Kiebooms G (2013). "Reproductive options for prospective parents in families with Huntington's disease: clinical, psychological and ethical reflections". Inson ko'payishining yangilanishi. 19 (3): 304–15. doi:10.1093/humupd/dms058. PMID 23377865.
- ^ Forrest Keenan K, Simpson SA, Miedzybrodzka Z, Alexander DA, Semper J (June 2013). "How do partners find out about the risk of Huntington's disease in couple relationships?". Journal of Genetic Counseling. 22 (3): 336–44. doi:10.1007/s10897-012-9562-2. PMID 23297124. S2CID 15447709.
- ^ Erwin C, Williams JK, Juhl AR, Mengeling M, Mills JA, Bombard Y, Hayden MR, Quaid K, Shoulson I, Taylor S, Paulsen JS (July 2010). "Perception, experience, and response to genetic discrimination in Huntington disease: the international RESPOND-HD study". Amerika tibbiyot genetikasi jurnali. Part B, Neuropsychiatric Genetics. 153B (5): 1081–93. doi:10.1002/ajmg.b.31079. PMC 3593716. PMID 20468061.
- ^ Burson CM, Markey KR (September 2001). "Genetic counseling issues in predictive genetic testing for familial adult-onset neurologic diseases". Bolalar nevrologiyasi bo'yicha seminarlar. 8 (3): 177–86. doi:10.1053/spen.2001.26451. PMID 11575847.
- ^ Smith JA, Michie S, Stephenson M, Quarrell O (March 2002). "Risk Perception and Decision-making Processes in Candidates for Genetic Testing for Huntington's Disease: An Interpretative Phenomenological Analysis". Sog'liqni saqlash psixologiyasi jurnali. 7 (2): 131–44. doi:10.1177/1359105302007002398. PMID 22114233. S2CID 40182214.
- ^ a b Hayden MR (March 2003). "Predictive testing for Huntington's disease: a universal model?". Lanset. Nevrologiya. 2 (3): 141–2. doi:10.1016/S1474-4422(03)00317-X. PMID 12849232. S2CID 39581496.
- ^ "Guidelines for the molecular genetics predictive test in Huntington's disease. International Huntington Association (IHA) and the World Federation of Neurology (WFN) Research Group on Huntington's Chorea". Nevrologiya. 44 (8): 1533–6. 1994 yil avgust. doi:10.1212/WNL.44.8.1533. PMID 8058167.
- ^ Losekoot M, van Belzen MJ, Seneca S, Bauer P, Stenhouse SA, Barton DE (May 2013). "EMQN/CMGS best practice guidelines for the molecular genetic testing of Huntington disease". Evropa inson genetikasi jurnali. 21 (5): 480–6. doi:10.1038/ejhg.2012.200. PMC 3641377. PMID 22990145.
- ^ Schulman JD, Black SH, Handyside A, Nance WE (February 1996). "Xantington kasalligi va ba'zi boshqa dominant irsiy kasalliklar uchun preimplantatsiya genetik tekshiruvi". Klinik genetika. 49 (2): 57–8. doi:10.1111 / j.1399-0004.1996.tb04327.x. PMID 8740912. S2CID 45703511.
- ^ Stern HJ, Harton GL, Sisson ME, Jones SL, Fallon LA, Thorsell LP, Getlinger ME, Black SH, Schulman JD (June 2002). "Non-disclosing preimplantation genetic diagnosis for Huntington disease". Prenatal diagnostika. 22 (6): 503–7. doi:10.1002/pd.359. PMID 12116316. S2CID 33967835.
- ^ "Predictive Testing for Huntington's Disease". 2011. Arxivlandi asl nusxasidan 2013 yil 22 yanvarda. Olingan 7 may 2013.
- ^ Kuliev A, Verlinsky Y (April 2005). "Preimplantation diagnosis: a realistic option for assisted reproduction and genetic practice". Akusherlik va ginekologiyaning hozirgi fikri. 17 (2): 179–83. doi:10.1097/01.gco.0000162189.76349.c5. PMID 15758612. S2CID 9382420.
- ^ "Guidelines for Genetic Testing for Huntington's Disease". Heredity Disease Foundation. Arxivlandi asl nusxasi 2015 yil 26 iyunda. Olingan 7 may 2013.
- ^ a b Schneider SA, Walker RH, Bhatia KP (September 2007). "The Huntington's disease-like syndromes: what to consider in patients with a negative Huntington's disease gene test". Tabiat klinikasi nevrologiyasi. 3 (9): 517–25. doi:10.1038/ncpneuro0606. PMID 17805246. S2CID 9052603.
- ^ Frank S, Jankovic J (March 2010). "Advances in the pharmacological management of Huntington's disease". Giyohvand moddalar. 70 (5): 561–71. doi:10.2165/11534430-000000000-00000. PMID 20329804. S2CID 42386743. Arxivlandi asl nusxasi on 8 October 2011.
- ^ a b v Bonelli RM, Wenning GK, Kapfhammer HP (March 2004). "Huntington's disease: present treatments and future therapeutic modalities". Xalqaro klinik psixofarmakologiya. 19 (2): 51–62. doi:10.1097/00004850-200403000-00001. PMID 15076012. S2CID 1956458.
- ^ Panagiotakis PH, DiSario JA, Hilden K, Ogara M, Fang JC (2008). "DPEJ tube placement prevents aspiration pneumonia in high-risk patients". Klinik amaliyotda ovqatlanish. 23 (2): 172–5. doi:10.1177/0884533608314537. PMID 18390785.
- ^ a b "EHDN Physiotherapy Guidance Document" (PDF). European HD Network Physiotherapy Working Group. Arxivlandi asl nusxasi (PDF) 2016 yil 4 martda. Olingan 15 noyabr 2015.
- ^ Quin L, Busee M (February 2012). "Development of physiotherapy guidance and treatment-based classifications for people with Huntington's disease". Neurodegenerative Disease Management. 2 (1): 21–31. doi:10.2217/nmt.11.86.
- ^ Khalil H, Quinn L, van Deursen R, Martin R, Rosser A, Busse M (January 2012). "Xantington kasalligi bo'lgan odamlarda uy sharoitida mashq qilish DVD-dan foydalanishga rioya qilish: ishtirokchilarning istiqbollari". Jismoniy terapiya. 92 (1): 69–82. doi:10.2522 / ptj.20100438. PMID 21960468.
- ^ Travers E, Jones K, Nicol J (2007). "Xantington kasalligida palyativ yordam ko'rsatish". Xalqaro palliativ hamshiralik jurnali. 13 (3): 125–30. doi:10.12968 / ijpn.2007.13.3.23274. PMID 17505405.
- ^ "FDA Xantington kasalligida xoreyani davolash uchun birinchi dori-darmonni ma'qulladi". AQSh oziq-ovqat va farmatsevtika idorasi. 2008 yil 15-avgust. Arxivlandi asl nusxasidan 2008 yil 21 avgustda. Olingan 10 avgust 2008.
- ^ Morsi S, Xalil SM, Dohaym MF, Kamel MG, El-Basioni DS, Ahmed Xasan HI va boshq. (Avgust 2019). "Xantington kasalligini davolash sifatida etil-EPA samaradorligi: tizimli tahlil va meta-tahlil". Acta Neuropsychiatrica. 31 (4): 175–185. doi:10.1017 / neu.2019.11. hdl:10069/39427. PMID 30890195. S2CID 84183892.
- ^ Tadqiqot, Giyohvand moddalarni baholash markazi (2019 yil 17-iyul). "Tardiv diskineziaga intilish: Valbenazinni belgilash va tasdiqlash". FDA. Olingan 15 noyabr 2020.
- ^ Sitrom L (2016 yil aprel). "Psixiatriya va nevrologiya o'rtasidagi aloqalar uchun yangi dorilar". Xalqaro klinik amaliyot jurnali. 70 (4): 298–9. doi:10.1111 / ijcp.12805. PMID 27028671. S2CID 38537781.
- ^ Harper P (2002). "Genetik maslahat va simptomatik tekshiruv". Bates G, Harper P, Jons L (tahrir). Xantington kasalligi - Uchinchi nashr. Oksford: Oksford universiteti matbuoti. 198-242 betlar. ISBN 978-0-19-851060-4.
- ^ Harper PS (iyun 1999). "Xantington kasalligi: poliglutamin takrorlanishining klinik, genetik va molekulyar modeli". London Qirollik Jamiyatining falsafiy operatsiyalari. B seriyasi, Biologiya fanlari. 354 (1386): 957–61. doi:10.1098 / rstb.1999.0446. PMC 1692597. PMID 10434293.
- ^ Endryu SE, Goldberg YP, Kremer B, Telenius H, Theilmann J, Adam S, Starr E, Squitieri F, Lin B, Kalchman MA (avgust 1993). "Trinukleotid (CAG) takroriy davomiyligi va Xantington kasalligining klinik xususiyatlari o'rtasidagi bog'liqlik". Tabiat genetikasi. 4 (4): 398–403. doi:10.1038 / ng0893-398. PMID 8401589. S2CID 20645822.
- ^ Krauford D, Snouden J (2002). "Xantington kasalligining neyropsixologik va neyropsikiyatrik jihatlari". Bates G, Harper P, Jons L (tahrir). Xantington kasalligi - Uchinchi nashr. Oksford: Oksford universiteti matbuoti. 62-87 betlar. ISBN 978-0-19-851060-4.
- ^ Di Maio L, Squitieri F, Napolitano G, Campanella G, Trofatter JA, Conneally PM (aprel 1993). "Xantington kasalligida o'z joniga qasd qilish xavfi". Tibbiy genetika jurnali. 30 (4): 293–5. doi:10.1136 / jmg.30.4.293. PMC 1016335. PMID 8487273.
- ^ a b v d e f Harper P (2002). "Xantington kasalligining epidemiologiyasi". Bates G, Harper P, Jons L (tahrir). Xantington kasalligi - Uchinchi nashr. Oksford: Oksford universiteti matbuoti. 159-189 betlar. ISBN 978-0-19-851060-4.
- ^ Sharon I, Sharon R, Wilkens JP, Ersan T (2010). "Xantington kasalligi demansi". tibbiyot, WebMD. Medscape. Arxivlandi asl nusxasidan 2010 yil 5 martda. Olingan 16 may 2010.
- ^ Drayv-Dankli E, Kavinis JN (2007). "Xantington kasalligi". Schapira AH (tahrir). Nevrologiya va klinik nevrologiya. Mosby Elsevier. 879-885 betlar. ISBN 978-0-323-03354-1.
- ^ Evans SJ, Duglas I, Rawlins MD, Veksler NS, Tabrizi SJ, Smit L (oktyabr 2013). "Buyuk Britaniyada kattalar Xantington kasalligining umumiy amaliyot yozuvlarida qayd etilgan tashxis asosida tarqalishi". Nevrologiya, neyroxirurgiya va psixiatriya jurnali. 84 (10): 1156–60. doi:10.1136 / jnnp-2012-304636. PMC 3786631. PMID 23482661.
- ^ Avila-Jiro R (1973). "Venesuela Zuliya shtatidagi Xantington xoreyasining tibbiy-ijtimoiy jihatlari". Nevrologiyaning yutuqlari. 1: 261–6. ISSN 0091-3952. NAID 10021247802.
- ^ a b Gusella JF, Wexler NS, Conneally PM, Naylor SL, Anderson MA, Tanzi RE, Watkins PC, Ottina K, Wallace MR, Sakaguchi AY (1983). "Hantington kasalligi bilan genetik bog'langan polimorf DNK markeri". Tabiat. 306 (5940): 234–8. Bibcode:1983 yil natur.306..234G. doi:10.1038 / 306234a0. PMID 6316146. S2CID 4320711.
- ^ Squitieri F, Andrew SE, Goldberg YP, Kremer B, Spence N, Zeisler J, Nichol K, Theilmann J, Greenberg J, Goto J (dekabr 1994). "Hantington kasalligining DNK-haplotipini tahlil qilish CAG kengayishining kelib chiqishi va mexanizmlari va tarqalishining geografik o'zgarishlari sabablari to'g'risida ma'lumotni ochib beradi". Inson molekulyar genetikasi. 3 (12): 2103–14. doi:10.1093 / hmg / 3.12.2103. PMID 7881406.
- ^ Sveinsson O, Halldorsson S, Olafsson E (iyul 2012). "Islandiyada Xantington kasalligining juda kam tarqalganligi". Evropa nevrologiyasi. 68 (1): 48–51. doi:10.1159/000337680. PMID 22722209. S2CID 207551998.
- ^ Sipilä JO, Hietala M, Siitonen A, Päivärinta M, Majamaa K (yanvar 2015). "Finlyandiyada Xantington kasalligi epidemiologiyasi". Parkinsonizm va unga aloqador buzilishlar. 21 (1): 46–9. doi:10.1016 / j.parkreldis.2014.10.025. PMID 25466405.
- ^ Almqvist EW, Elterman DS, MacLeod PM, Hayden MR (sentyabr 2001). "Britan Kolumbiyasida Xantington kasalligiga yangi tashxis qo'yilgan bemorlarning to'rtdan birida kasallikning yuqori darajasi va oilaning yo'qligi". Klinik genetika. 60 (3): 198–205. doi:10.1034 / j.1399-0004.2001.600305.x. PMID 11595021. S2CID 19786394.
- ^ a b Xantington G (1872). Xoreada. Filadelfiyaning tibbiy va jarrohlik bo'yicha muxbiri. 26. Gaaga: Nixoff. pp.317 –321. ISBN 978-90-6186-011-2. Olingan 1 aprel 2009.
- ^ Bellenir K, ed. (2004). "Xantington kasalligi". Genetik kasalliklar haqida ma'lumot (3-nashr). Detroyt: Omnigrafika. pp.159 –179. ISBN 978-0-7808-0742-6.
- ^ a b v d e f g h men j Harper P (2002). "Xantington kasalligi: tarixiy zamin". Bates G, Harper P, Jons L (tahrir). Xantington kasalligi - Uchinchi nashr. Oksford: Oksford universiteti matbuoti. 3-24 betlar. ISBN 978-0-19-851060-4.
- ^ a b v d Veksler A, Veksler N (2008). Dengizga yurgan ayol. Xantington va genetik kasallikning paydo bo'lishi. Yel universiteti matbuoti. p. 288. ISBN 978-0-300-10502-5. Olingan 15 noyabr 2015.
- ^ Lund JK (1860). "Chorea Sti Viti i Sætersdalen. Uddrag af Distriktslæge J. C. Lunds Medicinalberetning for 1860". Om Sundhedstilstanden-ga murojaat qiling: 137–138.
- ^ Lanska DJ (aprel 2000). "Jorj Xantington (1850–1916) va irsiy xoreya". Neuroscience tarixi jurnali. 9 (1): 76–89. doi:10.1076 / 0964-704X (200004) 9: 1; 1-2; FT076. PMID 11232352. S2CID 22659368.
- ^ Brodi IA, Uilkins RH (1967 yil sentyabr). "Xantingtonning xoresi". Nevrologiya arxivi. 17 (3): 331. doi:10.1001 / archneur.1967.00470270109013. PMID 4228262.
- ^ Jelliffe SE, Muncey EB, Davenport CB (1913). "Xantington xoreyasi: nasl-nasabda o'rganish". Asab va ruhiy kasalliklar jurnali. 40 (12): 796–799. doi:10.1097/00005053-191312000-00010.
- ^ a b Davenport CB, Muncey EB (1916). "Hentingtonning xoreyasi irsiyat va evgenikaga nisbatan". Amerika jinnilik jurnali. 73 (2): 195–222. doi:10.1176 / ajp.73.2.195.
- ^ Vessi PR (1932). "300 yil davomida Xantington xorasini yuqtirish to'g'risida - Bures oilaviy guruhi". Asab va ruhiy kasallik. 76 (6): 553–573. doi:10.1097/00005053-193212000-00001. S2CID 147656032. Olingan 1 aprel 2009.
- ^ a b Wexler AR (2002). "XIX asrdagi shaharda xorea va jamoa". Tibbiyot tarixi byulleteni. 76 (3): 495–527. doi:10.1353 / bhm.2002.0150. PMID 12486915. S2CID 30791504.
- ^ Conneally PM (may 1984). "Xantington kasalligi: genetika va epidemiologiya". Amerika inson genetikasi jurnali. 36 (3): 506–26. PMC 1684448. PMID 6233902.
- ^ Wexler NS (2012). "Xantington kasalligi: ilm-fanni targ'ib qilish". Tibbiyotning yillik sharhi. 63: 1–22. doi:10.1146 / annurev-med-050710-134457. PMID 22248319.
- ^ a b "Venesuela Xantington kasalligi loyihasi". Herediter Disease Foundation veb-sayti. Herediter Disease Foundation. 2008. Arxivlangan asl nusxasi 2015 yil 10-avgustda. Olingan 8 sentyabr 2008.
- ^ a b Macdonald M (mart 1993). "Trinukleotid takrorlanishini o'z ichiga olgan yangi gen, Xantington kasalligi xromosomalarida kengaygan va beqaror. Xantington kasalliklari bo'yicha hamkorlik tadqiqot guruhi". Hujayra. 72 (6): 971–83. doi:10.1016 / 0092-8674 (93) 90585-E. hdl:2027.42/30901. PMID 8458085. S2CID 802885.
- ^ Bertram L, Tanzi RE (iyun 2005). "Neyrodejenerativ kasallikning genetik epidemiologiyasi". Klinik tadqiqotlar jurnali. 115 (6): 1449–57. doi:10.1172 / JCI24761. PMC 1137006. PMID 15931380.
- ^ La Spada AR, Rolling DB, Harding AE, Warner CL, Spiegel R, Hausmanova-Petrusevich I, Yee WC, Fisbeck KH (1992 yil dekabr). "X-ga bog'liq o'murtqa va bulbar mushak atrofiyasida trinukleotid takrorlanishining meiotik barqarorligi va genotip-fenotip korrelyatsiyasi". Tabiat genetikasi. 2 (4): 301–4. doi:10.1038 / ng1292-301. PMID 1303283. S2CID 6603129.
- ^ a b v Ross CA, Tabrizi SJ (yanvar 2011). "Xantington kasalligi: molekulyar patogenezdan klinik davolashgacha". Lanset. Nevrologiya. 10 (1): 83–98. doi:10.1016 / S1474-4422 (10) 70245-3. PMID 21163446. S2CID 17488174.
- ^ "HD nima?". Xantington kasalliklari assotsiatsiyasi. Arxivlandi asl nusxasi 2011 yil 13 dekabrda. Olingan 18 dekabr 2011.
- ^ Davenport CB (1915 yil may). "Hentingtonning xoreyasi irsiyat va evgenika bilan bog'liq". Amerika Qo'shma Shtatlari Milliy Fanlar Akademiyasi materiallari. 1 (5): 283–5. Bibcode:1915PNAS .... 1..283D. doi:10.1073 / pnas.1.5.283. PMC 1090803. PMID 16575999.
- ^ Rollin BE (2006). "Hayvonlarni tadqiq qilishni tartibga solish va hayvon axloqining paydo bo'lishi: kontseptual tarix" (PDF). Nazariy tibbiyot va bioetika. 27 (4): 285–304. doi:10.1007 / s11017-006-9007-8. PMID 16937023. S2CID 18620094.
- ^ Doerflinger RM (2008 yil fevral). "Embrional ildiz hujayralarini tadqiq qilishda aldanish muammosi". Hujayraning tarqalishi. 41 (Qo'shimcha 1): 65-70. doi:10.1111 / j.1365-2184.2008.00492.x. PMC 6496399. PMID 18181947.
- ^ Chapman MA (1990 yil iyul). "Voyaga etgan genetik kasallik uchun bashoratli sinov: Xantington kasalligi uchun bog'lanish tahlilidan foydalanishning axloqiy va huquqiy oqibatlari". Amerika inson genetikasi jurnali. 47 (1): 1–3. PMC 1683745. PMID 2140926.
- ^ Xuggins M, Bloch M, Kanani S, Quarrell OW, Theilman J, Hedrik A, Dikkens B, Linch A, Xayden M (iyul 1990). "Voyaga etgan kasallikni prognozli tekshirishda paydo bo'lgan axloqiy va huquqiy muammolar: Xantington kasalligi tajribasi". Amerika inson genetikasi jurnali. 47 (1): 4–12. PMC 1683755. PMID 1971997.
- ^ "Sug'urta genetikasi bo'yicha moratoriy 2017 yilgacha uzaytirildi" (Matbuot xabari). Britaniya sug'urtachilari assotsiatsiyasi. 2011 yil 5 aprel. Arxivlandi asl nusxasidan 2016 yil 4 martda. Olingan 13 yanvar 2016.
- ^ "Mutaxassis gen testini oshkor qilishni qo'llab-quvvatlaydi". BBC maqolasi. 2007 yil 7-iyun. Arxivlandi asl nusxasidan 2008 yil 26 fevralda.
- ^ Binedell J, Soldan JR, Scourfield J, Harper PS (noyabr 1996). "Xantington kasalligining prognozli tekshiruvi: o'spirinlarning so'rovlariga baholash yondashuvi uchun masala". Tibbiy genetika jurnali. 33 (11): 912–8. doi:10.1136 / jmg.33.11.912. PMC 1050784. PMID 8950670.
- ^ Borry P, Goffin T, Nys H, Dierickx K (2008). "Voyaga etmaganlarda voyaga etgan genetik kasalliklar bo'yicha prognozli genetik tekshiruv". Sinay tog'idagi tibbiyot jurnali, Nyu-York. 75 (3): 287–96. doi:10.1002 / msj.20038. PMID 18704981.
- ^ a b v Braude PR, De Wert GM, Evers-Kiebooms G, Pettigrew RA, Geraedts JP (1998 yil dekabr). "Xantington kasalligi uchun preplantatsiya preimplantatsiyasining genetik diagnostikasi: amaliy va axloqiy dilemmalar". Prenatal diagnostika. 18 (13): 1422–6. doi:10.1002 / (SICI) 1097-0223 (199812) 18:13 <1422 :: AID-PD499> 3.0.CO; 2-R. PMID 9949442.
- ^ "Xalqaro Xantington uyushmasi". Xalqaro Xantington uyushmasi. 2013 yil. Arxivlandi asl nusxasidan 2009 yil 18 aprelda. Olingan 3 aprel 2009.
- ^ "AQSh Senatining 531-sonli qarori".. S. Res. 531. AQSh Senati. 6 aprel 2008 yil. Arxivlandi asl nusxasidan 2015 yil 17 noyabrda. Olingan 10 avgust 2008.
- ^ a b Odling-Smee L (2007 yil 17-may). "Biyomedikal xayriya: Pul daraxti". Tabiat. 447 (7142): 251. Bibcode:2007 yil natur.447..251.. doi:10.1038 / 447251a. S2CID 4357517.
- ^ a b "CHDI Foundation". chdifoundation.org. Olingan 13 noyabr 2020.
- ^ E ni tekshiring (2007 yil may). "Biyomedikal xayriya: sevgi yoki pul". Tabiat. 447 (7142): 252–3. Bibcode:2007 yil natur.447..252C. doi:10.1038 / 447252a. PMID 17507955.
- ^ a b "Xantington kasalligi: tadqiqotlar orqali umid". www.ninds.nih.gov. Olingan 16 noyabr 2020.
- ^ Tyorner S, Schapira AH (iyun 2010). "Miyaning mitoxondriyal masalalari: Xantington kasalligidagi roli". Bioenergetika va biomembranalar jurnali. 42 (3): 193–8. doi:10.1007 / s10863-010-9290-y. PMID 20480217. S2CID 207185615.
- ^ Wild EJ, Tabrizi SJ (sentyabr 2014). "Xantington kasalligi bo'yicha kelgusidagi klinik sinovlarning maqsadlari: bu nimani anglatadi?". Harakatning buzilishi. 29 (11): 1434–45. doi:10.1002 / mds.26007. PMC 4265300. PMID 25155142.
- ^ a b Sulaymon, Shoshanna (2017 yil 3-may). "Taube AQShda Xantington kasalligi bo'yicha tadqiqotlarni moliyalashtiradi". The Times of Israel. Arxivlandi asl nusxasidan 2017 yil 3 mayda. Olingan 5 may 2017.
- ^ a b Munoz-Sanjuan I, Bates GP (2011 yil fevral). "Xantington kasalligi uchun yangi davolash usullarini ishlab chiqishda asosiy va klinik tadqiqotlarni birlashtirishning ahamiyati". Klinik tadqiqotlar jurnali. 121 (2): 476–83. doi:10.1172 / JCI45364. PMC 3026740. PMID 21285520.
- ^ McBride JL, Pitser MR, Boudreau RL, Dyufour B, Hobbs T, Ojeda SR, Davidson BL (dekabr 2011). "Xantington kasalligi uchun potentsial terapiya sifatida rezus makakasida RNAi vositachiligida HTT bostirilishining klinikgacha xavfsizligi". Molekulyar terapiya. 19 (12): 2152–62. doi:10.1038 / mt.2011.219. PMC 3242667. PMID 22031240.
- ^ Kordasiewicz HB, Stanek LM, Wancewicz EV, Mazur C, McAlonis MM, Pytel KA, Artates JW, Vayss A, Cheng SH, Shihabuddin LS, Hung G, Bennett CF, Klivlend DW (iyun 2012). "Hantington sintezining vaqtinchalik repressiyasi bilan Xantington kasalligini barqaror terapevtik qayta tiklash". Neyron. 74 (6): 1031–44. doi:10.1016 / j.neuron.2012.05.009. PMC 3383626. PMID 22726834.
- ^ Barns DW, Whitley RJ (fevral 1987). "Antiviral terapiya va o'pka kasalligi". Ko'krak qafasi. 91 (2): 246–51. doi:10.1172 / JCI45130. PMC 3026739. PMID 3026739.
- ^ "Landmark Huntingtonning sud jarayoni boshlandi". Arxivlandi asl nusxasidan 2015 yil 21 oktyabrda. Olingan 19 oktyabr 2015.
- ^ "Xantington kasalligi erta bo'lgan bemorlarda IONIS-HTTRx xavfsizligi, bardoshliligi, farmakokinetikasi va farmakodinamikasi - to'liq matnli ko'rinish". ClinicalTrials.gov. Arxivlandi asl nusxasidan 2015 yil 29 sentyabrda. Olingan 18 aprel 2016.
- ^ Wild EJ, Boggio R, Langbehn D, Robertson N, Haider S, Miller JR, Zetterberg H, Leavitt BR, Kuhn R, Tabrizi SJ, Macdonald D, Vayss A (may 2015). "Xantington kasalligi bilan og'rigan bemorlarda miya omurilik suyuqligidagi mutant huntin oqsilining miqdori". Klinik tadqiqotlar jurnali. 125 (5): 1979–86. doi:10.1172 / jci80743. PMC 4463213. PMID 25844897.
- ^ Chase A (2015 yil may). "Xantington kasalligi: prodromal HD uchun miya omurilik suyuqligi va MRI biomarkerlari". Tabiat sharhlari Nevrologiya. 11 (5): 245. doi:10.1038 / nrneurol.2015.63. PMID 25896083. S2CID 38300571.
- ^ Keizer MS, Kordasiewicz HB, McBride JL (2016 yil aprel). "Dominant naslga o'tgan neyrodejenerativ kasalliklar uchun genlarni to'xtatish strategiyalari: Xantington kasalligi va spinoserebellar ataksiya". Inson molekulyar genetikasi. 25 (R1): R53-64. doi:10.1093 / hmg / ddv442. PMC 4802374. PMID 26503961.
- ^ a b Djajadikerta, Alvin; Keshri, Svati; Pavel, Mariana; Prestil, Rayan; Rayan, Laura; Rubinsztein, David C. (3 aprel 2020). "Nörodejeneratif kasalliklar uchun terapevtik strategiya sifatida otofagiya induksiyasi". Molekulyar biologiya jurnali. Neyrodejenerativ kasalliklarda avtofagiya. 432 (8): 2799–2821. doi:10.1016 / j.jmb.2019.12.035. ISSN 0022-2836. PMID 31887286.
- ^ Ravikumar, Brinda; Vaxer, Korali; Berger, Zdenek; Devies, Janet E.; Luo, Shouking; Oroz, Lourdes G.; Skaravilli, Franchesko; Iston, Duglas F.; Dyuden, Rayner; O'Kane, Cahir J.; Rubinsztein, Devid C. (2004 yil iyun). "MTORni inhibe qilish avtofagiyani keltirib chiqaradi va Hantington kasalligining chivin va sichqon modellarida poliglutamin kengayishining toksikligini pasaytiradi". Tabiat genetikasi. 36 (6): 585–595. doi:10.1038 / ng1362. ISSN 1546-1718. PMID 15146184. S2CID 7749825.
- ^ Clelland CD, Barker RA, Watts C (2008). "Xantington kasalligida hujayra terapiyasi". Neyroxirurgik diqqat. 24 (3-4): E9. doi:10.3171 / FOC / 2008/24 / 3-4 / E8. PMID 18341412.
- ^ Kundiff PE, Anderson SA (iyun 2011). "Induktsiyalangan pluripotent ildiz hujayralarining markaziy asab tizimi kasalliklarini o'rganishga ta'siri". Genetika va rivojlanish sohasidagi dolzarb fikrlar. 21 (3): 354–61. doi:10.1016 / j.gde.2011.01.008. PMC 3932563. PMID 21277194.
- ^ "Qidiruv: Xantington kasalligi - ro'yxat natijalari - ClinicalTrials.gov". kliniktrials.gov.
- ^ "Tugallangan klinik sinovlar". Huntington Study Group. Arxivlandi asl nusxasi 2012 yil 28 iyunda. Olingan 4 fevral 2012.
Tashqi havolalar
- Xantington kasalligi da Curlie
- HOPES loyihasi – Stenford universitetining HD ma'lumot loyihasi
- HDBuzz - Olimlar tomonidan oddiy tilda yozilgan HD tadqiqot yangiliklari
- HD dori vositalari - joriy muolajalar va rejalashtirilgan sinovlar haqidagi yangiliklar
| Tasnifi | |
|---|---|
| Tashqi manbalar |